
A homozygous nonsense mutation in SOX9 in the dominant disorder campomelic dysplasia: a case of...
11
ORIGINAL INVESTIGATION Ramona Pop Michael V. Zaragoza Mara Gaudette Ulrike Dohrmann Gerd Scherer A homozygous nonsense mutation in SOX9 in the dominant disorder campomelic dysplasia: a case of mitotic gene conversion Received: 16 December 2004 / Accepted: 6 February 2005 / Published online: 2 April 2005 ȑ Springer-Verlag 2005 Abstract Campomelic dysplasia (CD; MIM 114290), an autosomal dominant skeletal malformation syndrome with XY sex reversal, is caused by heterozygous de novo mutations in and around the SOX9 gene on 17q. We report a patient with typical signs of CD, including sex reversal, who was, surprisingly, homozygous for the nonsense mutation Y440X. Since neither parent carried the Y440X mutation, possible mechanisms explaining the homozygous situation were a de novo mutation followed by uniparental isodisomy, somatic crossing over, or gene conversion. As the patient was heterozy- gous for six microsatellite markers flanking SOX9, uni- parental isodisomy and somatic crossing over were excluded. Analysis of intragenic single-nucleotide poly- morphisms suggested that the homozygous mutation arose by a mitotic gene conversion event involving ex- change of at least 440 nucleotides and at most 2,208 nucleotides between a de novo mutant maternal allele and a wild-type paternal allele. Analysis of cloned alleles showed that homozygous mutant cells constituted about 80% of the leukocyte cell population of the patient, whereas about 20% were heterozygous mutant cells. Heterozygous Y440X mutations, previously described in three CD cases, have been identified in seven additional cases, thus constituting the most frequent recurrent mutations in SOX9. These patients frequently have a milder phenotype with longer survival, possibly because of the retention of some transactivation activity of the mutant protein on SOX9 target genes, as shown by cell transfection experiments. The fact that the patient sur- vived for 3 months may thus be explained by homozy- gosity for a hypomorphic rather than a complete loss-of- function allele, in combination with somatic mosaicism. This is, to our knowledge, the first report of mitotic gene conversion of a wild-type allele by a de novo mutant allele in humans. Introduction Campomelic dysplasia (CD; MIM 114290), an autoso- mal dominant skeletal malformation syndrome often accompanied by XY sex reversal, is caused by hetero- zygous de novo mutations in and around the SOX9 gene at 17q. The hallmark symptoms of the fully manifested disease seen in the majority of patients are skeletal abnormalities including bowing of femora and tibiae, hypoplastic scapulae, 11 instead of 12 pairs of ribs, nonmineralized thoracic pedicles, pelvis and spine mal- formations, Robin sequence, and bilateral clubfoot (Houston et al. 1983; Mansour et al. 1995). The absence of olfactory tracts and bulbs, and heart and renal mal- formations have been noted. Most patients die in the neonatal period because of respiratory distress, which has been attributed to hypoplasia of the tracheobron- chial cartilage and small thoracic cage. Three-quarters of XY males have autosomal sex reversal with partial or R. Pop U. Dohrmann G. Scherer (&) Institute of Human Genetics and Anthropology, University of Freiburg, Breisacherstrasse 33, 79106 Freiburg, Germany E-mail: [email protected] Tel.: +49-761-270-7030 Fax: +49-761-270-7041 R. Pop Faculty for Biology, University of Freiburg, Freiburg, Germany M. V. Zaragoza M. Gaudette Division of Human Genetics, Department of Pediatrics, University of California Irvine, Orange, Calif., USA Present address: R. Pop Division of Hematology/Oncology, Department of Cancer Biology/Pediatrics, University of Massachusetts Medical School, Worcester, Mass., USA Hum Genet (2005) 117: 43–53 DOI 10.1007/s00439-005-1295-y
-
Upload
ramona-pop -
Category
Documents
-
view
214 -
download
2
Transcript of A homozygous nonsense mutation in SOX9 in the dominant disorder campomelic dysplasia: a case of...

ORIGINAL INVESTIGATION
Ramona Pop Æ Michael V. Zaragoza Æ Mara Gaudette
Ulrike Dohrmann Æ Gerd Scherer
A homozygous nonsense mutation in SOX9 in the dominant disordercampomelic dysplasia: a case of mitotic gene conversion
Received: 16 December 2004 / Accepted: 6 February 2005 / Published online: 2 April 2005� Springer-Verlag 2005
Abstract Campomelic dysplasia (CD; MIM 114290), anautosomal dominant skeletal malformation syndromewith XY sex reversal, is caused by heterozygous de novomutations in and around the SOX9 gene on 17q. Wereport a patient with typical signs of CD, including sexreversal, who was, surprisingly, homozygous for thenonsense mutation Y440X. Since neither parent carriedthe Y440X mutation, possible mechanisms explainingthe homozygous situation were a de novo mutationfollowed by uniparental isodisomy, somatic crossingover, or gene conversion. As the patient was heterozy-gous for six microsatellite markers flanking SOX9, uni-parental isodisomy and somatic crossing over wereexcluded. Analysis of intragenic single-nucleotide poly-morphisms suggested that the homozygous mutationarose by a mitotic gene conversion event involving ex-change of at least 440 nucleotides and at most 2,208nucleotides between a de novo mutant maternal allele
and a wild-type paternal allele. Analysis of cloned allelesshowed that homozygous mutant cells constituted about80% of the leukocyte cell population of the patient,whereas about 20% were heterozygous mutant cells.Heterozygous Y440X mutations, previously described inthree CD cases, have been identified in seven additionalcases, thus constituting the most frequent recurrentmutations in SOX9. These patients frequently have amilder phenotype with longer survival, possibly becauseof the retention of some transactivation activity of themutant protein on SOX9 target genes, as shown by celltransfection experiments. The fact that the patient sur-vived for 3 months may thus be explained by homozy-gosity for a hypomorphic rather than a complete loss-of-function allele, in combination with somatic mosaicism.This is, to our knowledge, the first report of mitotic geneconversion of a wild-type allele by a de novo mutantallele in humans.
Introduction
Campomelic dysplasia (CD; MIM 114290), an autoso-mal dominant skeletal malformation syndrome oftenaccompanied by XY sex reversal, is caused by hetero-zygous de novo mutations in and around the SOX9 geneat 17q. The hallmark symptoms of the fully manifesteddisease seen in the majority of patients are skeletalabnormalities including bowing of femora and tibiae,hypoplastic scapulae, 11 instead of 12 pairs of ribs,nonmineralized thoracic pedicles, pelvis and spine mal-formations, Robin sequence, and bilateral clubfoot(Houston et al. 1983; Mansour et al. 1995). The absenceof olfactory tracts and bulbs, and heart and renal mal-formations have been noted. Most patients die in theneonatal period because of respiratory distress, whichhas been attributed to hypoplasia of the tracheobron-chial cartilage and small thoracic cage. Three-quarters ofXY males have autosomal sex reversal with partial or
R. Pop Æ U. Dohrmann Æ G. Scherer (&)Institute of Human Genetics and Anthropology,University of Freiburg,Breisacherstrasse 33,79106 Freiburg,GermanyE-mail: [email protected].: +49-761-270-7030Fax: +49-761-270-7041
R. PopFaculty for Biology,University of Freiburg,Freiburg, Germany
M. V. Zaragoza Æ M. GaudetteDivision of Human Genetics,Department of Pediatrics,University of California Irvine,Orange, Calif., USA
Present address: R. PopDivision of Hematology/Oncology,Department of Cancer Biology/Pediatrics,University of Massachusetts Medical School,Worcester, Mass., USA
Hum Genet (2005) 117: 43–53DOI 10.1007/s00439-005-1295-y

complete gonadal dysgenesis (Houston et al. 1983;Mansour et al. 1995). Since all CD mutations have beenfound on only one allele of SOX9 with the other allelebeing unaffected, haplo-insufficiency for SOX9 is themost probable cause of the CD and sex reversal phe-notypes (Foster et al. 1994; Wagner et al. 1994).
SOX9 is a member of the SOX (SRY-type HMG box)family of transcription factors. Members of this familyare characterized by a related 79-amino acid DNA-binding motif known as the high-mobility group (HMG)domain. SOX9 also contains a transactivation domainlocated at the C-terminus of the protein (Sudbeck et al.1996; McDowall et al. 1999). Recently, a conserveddomain preceding the HMG domain has been identifiedthat is required for DNA-dependent dimerization ofSOX9 at paired binding sites within target promoters(Bernard et al. 2003; Sock et al. 2003). In accordancewith the CD phenotype, SOX9 is expressed throughoutchondrogenesis and acts as a key regulator at multiplesteps of cartilage differentiation (Akiyama et al. 2002).SOX9 is also expressed in the genital ridges of bothsexes, becoming upregulated in the developing testis anddownregulated in the developing ovary, consistent witha role in male sex determination (Kent et al. 1996;Morais da Silva et al. 1996). Direct evidence for this rolehas been provided by ectopic expression of Sox9 in XXtransgenic mice, which develop testes instead of ovaries(Vidal et al. 2001). Furthermore, SOX9 has been shownto bind to the promoters or enhancers of several chon-drocyte and testis differentiation genes such as the col-lagen genes Col2a1 and Col11a2 (Lefebvre et al. 1997;Ng et al. 1997; Bridgewater et al. 1998) and the testicularAmh gene (De Santa Barbara et al. 1998; Arango et al.1999).
SOX9 mutations in CD occur across the entire openreading frame and include missense, nonsense, frame-shift, and splice mutations. Nonsense mutations lead totruncated SOX9 proteins that miss part or all of thetransactivation domain, suggesting that, in these cases,impairment of gonadal and skeletal development resultsfrom the failure of target gene activation (Sudbeck et al.1996). CD has also been associated with a number ofchromosome rearrangements involving 17q, such as re-ciprocal translocations, inversions, or deletions (Pfeiferet al. 1999; Pop et al. 2004). Even though many molec-ular defects within the SOX9 gene have been reported,no distinct genotype/phenotype correlation concerningmutation type or position and severity of the disease andassociated sex reversal is apparent (Foster et al. 1994;Wagner et al. 1994; Meyer et al. 1997), pointing toincomplete penetrance or variable expressivity of thedisease.
In the present study, we describe a CD patient whowas found to be homozygous for the nonsense mutationY440X. We provide evidence that this homozygousmutation was the result of a nonreciprocal transfer ofpart of one de novo mutant maternal allele to the nor-mal paternal allele by mitotic gene conversion. The pa-tient also had somatic mosaicism. As has been shown
previously, Y440X is one of the few recurrent mutationsthat seems to be correlated with a longer survival time(Meyer et al. 1997) and, as we report here, is a muta-tional hot spot in the SOX9 gene. We demonstrate thefunctional consequences of the Y440X mutant proteinon genuine SOX9 target genes during bone and testisdevelopment and show that the mutant protein retainssome transactivation activity. The data suggest that thisis a hypomorphic rather than a complete loss-of-func-tion allele and, in concert with somatic mosaicism, ac-counted for the 3-month survival of the patient.
Materials and methods
Case report
Patient AG was the first-born child of a nonconsangu-inous and phenotypically normal Mexican couple. Ped-igree analysis was noncontributory. Prenatal ultrasoundat 19 weeks gestation revealed what appeared to be aright femur fracture and either a bent or fractured leftfemur, bilateral short tibiae and fibulae, and clubfeet.Follow-up ultrasounds demonstrated no demineraliza-tion or rib fractures and normal upper limbs. The babywas born at 39 weeks gestation by Cesarean section be-cause of the concern for fractures, which however werenot confirmed after birth. Examination revealed typicalclinical features and radiological features of CD. She hada large head, short thorax, short and bowed lower limbswith the characteristic pretibial dimples, and clubfeet.Her face had a flat nasal base and small chin (Fig. 1a).Radiological examination revealed a bell-shaped thorax,poorly mineralized thoracic pedicles, and hypoplasticscapulae (Fig. 1b, left), and narrow iliac wings and bentfemora and tibiae (Fig. 1b, right). She was phenotypi-cally female with a 46,XY karyotype, consistent with sexreversal. Additional studies showed hearing loss andnormally formed kidneys, heart, and brain. During herhospital stay, she had multiple complications includingfeeding difficulties because of pyloric stenosis. At age 5weeks, she was sent home on oxygen and with hospicecare. She died at the age of 3 months because of pro-gressive respiratory compromise.
Polymerase chain reaction, restriction,and sequence analyses
Genomic DNA was isolated from peripheral blood fromthe proband and her parents by standard techniques.The three exons and the exon/intron boundaries ofSOX9 were amplified by polymerase chain reaction(PCR) as described (Meyer et al. 1997) and used directlyfor DNA sequencing. The single-nucleotide polymor-phisms (SNPs) in intron 2 and in the 3¢-untranslatedregion were analyzed by sequencing and DdeI restriction
44

analysis, respectively, of PCR products cloned into thepCRII TOPO cloning vector by using the TOPO TACloning Kit (Invitrogen). Primers used were In2F: 5¢-actcgg gtg agt cgc ccc tcg a-3¢, In2R: 5¢-gat tgc cct gtg gacaat aaa-3¢, 3¢SNPsf: 5¢-gct taa ttc ctc agg ctt tgc g-3¢,3¢SNPsr: 5¢-tga cag agc gag cag gca tc-3¢. The 237-bpPCR product spanning codon 440 that was analyzed bydigestion with RsaI was amplified with primers Ex3F:5¢-cga acg cac atc aag acg ga-3¢ and Ex3R: 5¢-agc ggg gttcat gta ggt ga-3¢. The 592-bp PCR product spanningcodon 440 and the two flanking SNPs was amplifiedwith primers Ex3F7(1): 5¢-atg tcc aag cag cag gcg-3¢ andEx3R5(1): 5¢-ggt cct ctc ttt ctt cgg-3¢, cloned, and ana-lyzed by RsaI restriction analysis and by sequencing.Sequence reactions were performed with the Thermo-sequenase II Dye Terminator Cycle Sequencing kit(Amersham Pharmacia) and analyzed on an ABI Prism
310 automated DNA sequencer (Applied Biosystems).Paternity was tested by using the PowerPlex 16 system(Promega) and Genotyper version 3.7 from AppliedBiosystems.
Microsatellite marker analysis
Polymorphic SOX9-flanking markers used in fragmentanalysis assays are listed in Table 1. Of severalpotentially polymorphic microsatellites close to SOX9tested, microsatellites STS12 and STS72 were infor-mative in thefamily and consisted of a complex repeatA12(TA)9(CA)21A2 and a (CA)16 repeat in the refer-ence genomic sequence, respectively. Primers flankingthese repeats were STS12F: 5¢-gta cca cac ctg cta tgccct atg c-3¢, STS12R: 5¢-cgc cac cat gcc tgg cta at-3¢,STS72F: 5¢-aac tgt tcc gga cct gag t-3¢, STS72R 5¢-gaagag gct gga ggc act-3¢. PCR with fluorescence-labeledforward andunlabeled reverse primers to amplify thesepolymorphic regions was performed as described (Popet al. 2004). Because of its complex nature, the STS12repeat region was analyzed by both fragment and se-quence analyses.
Fig. 1 Clinical and radiological findings in patient AG. a Clinicalfeatures: macrocephaly, flat nasal base, small chin, and shortthorax. Note short bowed lower limbs with the characteristicpretibial dimples and clubfeet. b Radiological features. Radiographof thorax (left) showing hypoplastic scapulae, nonmineralizedthoracic pedicles, and 11 pairs of ribs. Radiograph of pelvis andlower extremities (right) showing vertically narrow iliac wings andbowed femora and tibiae
45

Quantitative PCR
Quantitative PCR analysis to measure gene dosage wasperformed by using a LightCycler and the FastStartDNA Master SYBR Green 1 Kit according to themanufacturer’s experimental protocol (Roche Diagnos-tics). The amount of genomic DNA per reaction was10 ng. Standard curves were generated with serially di-luted DNA from a normal individual. DNA from a CDpatient with one deleted SOX9 allele was used as acontrol (Pop et al. 2004). Samples to which water insteadof DNA was added served as negative controls. ThePCR cycle parameters were: 10 min at 95�C, followed by35 cycles of 95�C for 15 s, 58�C for 10 s, and 72�C for20 s. Subsequently, a melting curve was applied. ParallelPCRs were performed with primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and tyrosineaminotransferase (TAT). Primers to amplify a 237-bpproduct spanning codon 440 of SOX9 were Ex3F andEx3R (see above). Primers to amplify a 156-bp fragmentfrom GAPDH were GAPDHf: 5¢-gta ttg ggc gcc tgg tcacca g-3¢ and GAPDHr: 5¢-ccg ttc tca gcc ttg acg gtg-3¢(positions 62 and 218, respectively, in accession no.BC023632). Primers to amplify a 184-bp fragment fromTAT were TATf: 5¢-gtc act gca ctc caa gtc cg-3¢ andTATr: 5¢-gct tca gag ctg gtg tct tg-3¢ (positions 3355 and3539, respectively, in accession no. X77471). At the endof the LightCycler runs, the PCR products were recov-ered, and their lengths were confirmed by agarose gelelectrophoresis. Data analysis was performed with thesecond derivative maximum method of the LightCyclersoftware. The relative concentrations of the SOX9amplicon were normalized to those of GAPDH andTAT. Each measurement was carried out in duplicate.
Plasmids
The pcDNA3-based expression vector for human SOX9with an aminoterminal FLAG epitope, SOX9-N-FLAG/1-509, was as described (Sudbeck et al. 1996).The mutant construct SOX9/1-440 was produced asfollows. First, a 592-bp PCR product (ranging fromnucleotides 1378 to 1970 of the SOX9 cDNA sequence;Wagner et al. 1994), amplified with primers Ex3F7(1)/Ex3R5(1) (see above) from the genomic DNA of thepatient, was cloned into the pCRII TOPO cloning vector(Invitrogen). From the resulting clone, an SfiI/PpuMIfragment (nucleotides 1424–1825) was transferred intoSfiI/PpuMI-cut SOX9-N-FLAG/1-509, resulting inSOX9/1-440. Construct SOX9/1-248 used as a controlwas produced by removal of an ApaI restriction frag-ment from SOX9-N-FLAG/1-509. All expression cas-settes were verified by DNA sequencing. The fireflyluciferase reporter plasmids used were: AMH luc con-taining the human AMH promoter from positons �154to +10 (de Santa Barbara et al. 1998; gift of F. Poulat);4·(DE) Col11a2 luc containing four copies of the DEenhancer from the mouse Col11a2 gene in front of the
Col2a1 minimal promoter (Bridgewater et al. 2003; giftof L. Bridgewater), and 4·48 Col2a1 p89luc containingfour copies of the mouse Col2a1 intronic enhancer infront of the Col2a1 minimal promoter (Lefebvre et al.1997; gift of B. de Crombrugghe).
Cell culture, transfection, and luciferase assays
Neuro2A cells were maintained in Dulbecco’s modifiedEagle’s medium (DMEM) supplemented with 10% fetalcalf serum. For transient transfections, cells were platedat 3.0·105 cells/well in 12-well tissue culture plates 24 hbefore transfection. FuGene6 transfection reagent wasused according to the manufacturer’s instructions(Roche Diagnostics). Endotoxin-free plasmid DNAswere prepared by using the EndoFree Plasmid Kit(Qiagen). Each well of cells was transfected with a Fu-Gene6-DNA mixture in 100 ll DMEM containing0.3 ll FuGene6 reagent and a total of 101 ng DNA,comprised of 50 ng pcDNA3 expression plasmid, 50 ngluciferase reporter plasmid, and 1 ng pPRL-SV40 Re-nilla luciferase plasmid (Promega). The mixture waspreincubated for 15 min before addition to the cellmonolayer. Cells were harvested for luciferase assays48 h post-transfection, by using the Dual LuciferaseAssay System, as described (Promega). Reporter geneactivities shown represent the average of three differentexperiments in triplicate culture and their SD values.Fold induction was determined by first normalizing eachfirefly luciferase value to the Renilla luciferase internalcontrol, averaging the normalized values, and dividingby the mean of the firefly reporter cotransfected withempty vector only.
Results
Homozygous de novo mutation Y440X
In a SOX9 mutational screening of CD patients with orwithout XY sex reversal, we unexpectedly identified ahomozygous mutation in patient AG diagnosed withCD (Fig. 1). She was phenotypically female with a46,XY karyotype and died at the age of 3 months be-cause of progressive respiratory compromise (see above,Case report). The homozygous mutation detected in thispatient occurred in codon 440 at nucleotide 1692, con-verting a tyrosine codon to a stop codon (Y440X;TAC fi TAA; c.1692C fi A; Fig. 2a). The mutationwas shown to be de novo by sequence analysis of thecorresponding region in DNA from the parents and bythe documentation of paternity through microsatellitemarker analysis (not shown). As the homozygousmutation was observed with different PCR primercombinations, a second mutation at a primer-bindingsite causing dropout of the wild-type allele in the patientsample could be excluded.
46

Y440X homozygous mutation resultsfrom gene conversion
Since neither parent carries the Y440X mutation, pos-sible mechanisms explaining the de novo homozygousmutation in the patient include uniparental isodisomy,somatic crossing over, deletion, or gene conversion.These possible mechanisms can be distinguished by the
patterns of marker losses. Initially, a panel of poly-morphic markers from the 17q24–q25 region containingthe SOX9 gene was evaluated. From these markers, sixwere fully informative (Table 1) and showed typicalMendelian inheritance with paternal and maternal al-leles being detected in the proposita. These data excludeuniparental isodisomy and also somatic crossing over asa possible cause for the homozygous mutation. MarkerSTS12 is 12 kb from SOX9 on the centromeric side,whereas marker STS72 is 72 kb away on the telomericside. Therefore, a deletion in the megabase size range, asin the SOX9 deletion cases known so far (Olney et al.1999; Pop et al. 2004), could be excluded. To rule outthat a small deletion in exon 3 simulated homozygosity
Fig. 2 SOX9 Y440X mutation in patient AG. a Patient AG ishomozygous for the Y440X nonsense mutation. The chromato-gram given right shows the homozygous C to A transversion atnucleotide position 1692 (asterisk) in the patient, resulting in theY440X stop codon indicated above. Note the low wild-type C peakbelow the mutant A peak, indicative of a mosaic situation. Thewild-type sequence from one parent is depicted left. b Patient AG ismosaic for mutant and wild-type SOX9 alleles. Left RsaI restrictionanalysis of 237-bp uncloned PCR products generated with primers2 + 3. The digested genomic PCR products from the mother (M),the father (F), the patient (P), and a patient heterozygous for theY440X mutation used as a control (C) are compared. PCRproducts from the parents are completely digested into subfrag-ments of 138 and 99 bp, consistent with the absence of themutation. Half of the PCR product from the control sampleremains uncut, whereas the majority of the PCR product remainsuncut in the patient sample, confirming her mosaic state (Ma 100-bp DNA ladder). Right Analysis of cloned 592-bp PCR productsfrom the patient. The PCR product, which spans the mutantnucleotide position 1692 and the flanking SNPs at position 1485and 1925 (Table 2), was amplified with primers 1 + 4, cloned andsubjected to RsaI restriction analysis and to sequence analysis. Of30 clones, 27 were derived from the mutant (M) allele and threefrom the normal paternal (N) allele. Primers used were: 1 Ex3F7, 2Ex3F, 3 Ex3R, 4 Ex3R5
Table 1 Microsatellite marker analysis in the AG family (allelesizes are in base pairs)
Marker Locationa Motherb Fatherb Patientb
D17S808 58025831–58026219 153 149 159 157 149 157D17S949 65976927–65977271 220 216 216 218 216 218STS12 67616362–67616556 276 276 256 256 276 256SOX9 67628775–67634156 – – – – – –STS72 67700604–67700781 177 175 175 169 175 169D17S1351 68183642–68183959 173 179 171 171 179 171D17S928 77846128–77846429 156 134 150 148 134 148
aLocation of the markers refers to the UCSC Human GenomeBrowser May 2004 Freeze (http://genome.ucsc.edu/)bMaternally and paternally transmitted haplotypes are given initalics and bold, respectively
47
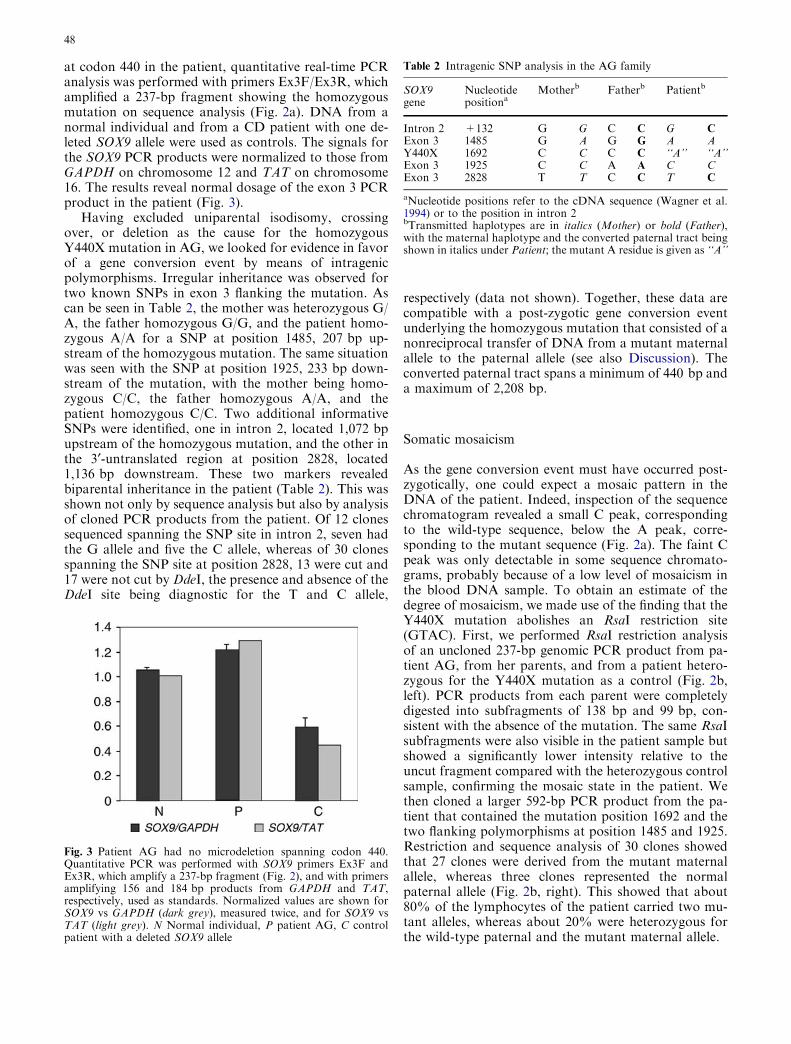
at codon 440 in the patient, quantitative real-time PCRanalysis was performed with primers Ex3F/Ex3R, whichamplified a 237-bp fragment showing the homozygousmutation on sequence analysis (Fig. 2a). DNA from anormal individual and from a CD patient with one de-leted SOX9 allele were used as controls. The signals forthe SOX9 PCR products were normalized to those fromGAPDH on chromosome 12 and TAT on chromosome16. The results reveal normal dosage of the exon 3 PCRproduct in the patient (Fig. 3).
Having excluded uniparental isodisomy, crossingover, or deletion as the cause for the homozygousY440X mutation in AG, we looked for evidence in favorof a gene conversion event by means of intragenicpolymorphisms. Irregular inheritance was observed fortwo known SNPs in exon 3 flanking the mutation. Ascan be seen in Table 2, the mother was heterozygous G/A, the father homozygous G/G, and the patient homo-zygous A/A for a SNP at position 1485, 207 bp up-stream of the homozygous mutation. The same situationwas seen with the SNP at position 1925, 233 bp down-stream of the mutation, with the mother being homo-zygous C/C, the father homozygous A/A, and thepatient homozygous C/C. Two additional informativeSNPs were identified, one in intron 2, located 1,072 bpupstream of the homozygous mutation, and the other inthe 3¢-untranslated region at position 2828, located1,136 bp downstream. These two markers revealedbiparental inheritance in the patient (Table 2). This wasshown not only by sequence analysis but also by analysisof cloned PCR products from the patient. Of 12 clonessequenced spanning the SNP site in intron 2, seven hadthe G allele and five the C allele, whereas of 30 clonesspanning the SNP site at position 2828, 13 were cut and17 were not cut by DdeI, the presence and absence of theDdeI site being diagnostic for the T and C allele,
respectively (data not shown). Together, these data arecompatible with a post-zygotic gene conversion eventunderlying the homozygous mutation that consisted of anonreciprocal transfer of DNA from a mutant maternalallele to the paternal allele (see also Discussion). Theconverted paternal tract spans a minimum of 440 bp anda maximum of 2,208 bp.
Somatic mosaicism
As the gene conversion event must have occurred post-zygotically, one could expect a mosaic pattern in theDNA of the patient. Indeed, inspection of the sequencechromatogram revealed a small C peak, correspondingto the wild-type sequence, below the A peak, corre-sponding to the mutant sequence (Fig. 2a). The faint Cpeak was only detectable in some sequence chromato-grams, probably because of a low level of mosaicism inthe blood DNA sample. To obtain an estimate of thedegree of mosaicism, we made use of the finding that theY440X mutation abolishes an RsaI restriction site(GTAC). First, we performed RsaI restriction analysisof an uncloned 237-bp genomic PCR product from pa-tient AG, from her parents, and from a patient hetero-zygous for the Y440X mutation as a control (Fig. 2b,left). PCR products from each parent were completelydigested into subfragments of 138 bp and 99 bp, con-sistent with the absence of the mutation. The same RsaIsubfragments were also visible in the patient sample butshowed a significantly lower intensity relative to theuncut fragment compared with the heterozygous controlsample, confirming the mosaic state in the patient. Wethen cloned a larger 592-bp PCR product from the pa-tient that contained the mutation position 1692 and thetwo flanking polymorphisms at position 1485 and 1925.Restriction and sequence analysis of 30 clones showedthat 27 clones were derived from the mutant maternalallele, whereas three clones represented the normalpaternal allele (Fig. 2b, right). This showed that about80% of the lymphocytes of the patient carried two mu-tant alleles, whereas about 20% were heterozygous forthe wild-type paternal and the mutant maternal allele.
Table 2 Intragenic SNP analysis in the AG family
SOX9gene
Nucleotidepositiona
Motherb Fatherb Patientb
Intron 2 +132 G G C C G CExon 3 1485 G A G G A AY440X 1692 C C C C ‘‘A’’ ‘‘A’’Exon 3 1925 C C A A C CExon 3 2828 T T C C T C
aNucleotide positions refer to the cDNA sequence (Wagner et al.1994) or to the position in intron 2bTransmitted haplotypes are in italics (Mother) or bold (Father),with the maternal haplotype and the converted paternal tract beingshown in italics under Patient; the mutant A residue is given as ‘‘A’’
Fig. 3 Patient AG had no microdeletion spanning codon 440.Quantitative PCR was performed with SOX9 primers Ex3F andEx3R, which amplify a 237-bp fragment (Fig. 2), and with primersamplifying 156 and 184 bp products from GAPDH and TAT,respectively, used as standards. Normalized values are shown forSOX9 vs GAPDH (dark grey), measured twice, and for SOX9 vsTAT (light grey). N Normal individual, P patient AG, C controlpatient with a deleted SOX9 allele
48

Mutant SOX9 protein retains some transactivationactivity
The Y440X mutation truncates the C-terminal transac-tivation (TA) domain spanning residues 402–509 ofSOX9 (Sudbeck et al. 1996). Previous studies haveshown residual transactivation potential for residues402–439 fused to a heterologous GAL4 DNA-bindingdomain when tested on a GAL4-dependent reporterplasmid (Meyer et al. 1997). We examined the effect ofthe truncated Y440X protein in its natural proteincontext by using authentic SOX9 target genes in tran-sient co-transfection experiments in Neuro2A cells. Full-length SOX9, 1-509, the Y440X mutant, 1-439, and atruncated form that lacked the entire TA domain, 1–248(Fig. 4a), were transfected with three different luciferasereporters. When four copies of the 48-bp Col2a1 intronicenhancer in front of the Col2a1 minimal promoter wereused as reporter (Lefebvre et al. 1997), we observed astrong response to full-length SOX9. This activation wasreduced to 5% when the 1–439 mutant was used,whereas the 1–248 control mutant showed negligibleactivation (Fig. 4b). Likewise, when four copies of theDE Col11a2 enhancer in front of the same Col2a1minimal promoter was used (Bridgewater et al. 2003), weobtained efficient activation by full-length SOX9, about
10% activation by the 1–439 mutant, and negligibleactivation by the 1–248 control (Fig. 4c). A higheractivation rate of about 22% of wild-type activity wasobtained when the 1–439 mutant was co-transfectedwith a reporter under control of the SOX9-responsiveAMH promoter (de Santa Barbara et al. 1998); here, theTA domain-lacking control 1–248 showed about 5%activation (Fig. 4d). Together, these data showed thatthe Y440X mutant retained some transactivationcapacity on authentic SOX9-responsive promoters/enh-ancers, ranging from 5% to 22% of wild-type activity.
Y440X is a SOX9 mutational hot spot
Three cases with a heterozygous Y440X mutation havebeen reported previously (Wagner et al. 1994; Meyeret al. 1997; Hageman et al. 1998). We have identifiedseven additional cases. The mutations consist of C fi Aor C fi G transversions, generating TAA or TAG stopcodons. Together, the recurrent Y440X mutation ac-counts for about 8% of all mutations in the SOX9protein-coding region (three of 37 published and sevenof our 90 unpublished cases). The karyotypes and sexualphenotypes of the ten patients with a heterozygousY440X mutation are listed in Table 3. Notably, many ofthese patients have a longer survival time compared tothe majority of CD patients, and several show only mildor no campomelia (see Discussion).
Discussion
We report a CD patient with a homozygous mutation inthe SOX9 gene who survived for 3 months after birth.This case is unique among all previously analyzed CDpatients. Homozygous mutations in dominant disordersare frequently embryonic lethals. Consistent with this,Sox9 null mice die during midgestation (Chaboissier
Fig. 4 Y440X truncated protein shows residual transactivationpotential. a Illustration of SOX9 full-length protein with itsfunctional domains and of its truncated constructs (D dimerizationdomain, HMG high mobility group domain, TA transactivationdomain). b–d Transactivation potential of SOX9 and of its deletionmutants determined by transient transfection into Neuro2A cells.Four copies of the 48-bp intronic enhancer of the Col2a1 gene(4·48 Col2a1 p89luc, b), four copies of the DE enhancer from theCol11a2 gene (4·(DE) Col11a2 p89luc, c), and the AMH promoter(AMH luc, d) were used to drive luciferase expression. Luciferasevalues obtained for each construct are given relative to the valueobtained for the vector alone, ±SD, from three differentexperiments, each performed in triplicates
49

et al. 2004). The fact that the patient survived for3 months despite having a homozygous mutation in theSOX9 gene was intriguing. We considered that a denovo mutation followed by somatic recombination ormitotic nondisjunction with reduplication of the mutantchromosome resulting in uniparental isodisomy mighthave been responsible for the loss of heterozygosity(LOH) in the patient. Some other more regionalizedevents such as gene conversion, deletion, or an addi-tional de novo mutation on the paternal chromosomecould also have occurred (Cavenee et al. 1993). Unipa-rental isodisomy, double crossing over, and a largedeletion were excluded by demonstrating heterozygosityfor microsatellite markers flanking the SOX9 gene.Furthermore, a small deletion in exon 3 of the paternalSOX9 allele was ruled out by quantitative PCR. Thus,the only remaining hypothesis was that a gene conver-sion event caused the homozygous mutation. This
implied that the patient should be homozygous forpolymorphic markers that immediately flank the muta-tion on either side and heterozygous for markers fartheraway. This was indeed found to be the case. These re-sults strongly imply that LOH is attributable to a non-reciprocal transfer that took place from the maternal tothe paternal allele, with the paternal converted tractspanning a minimum of 440 bp and a maximum of2,208 bp. The interpretation of our result is shownschematically in Fig. 5. We conclude that the homozy-gous mutation arose in a two-step manner: first, a denovo germline mutation occurred on the maternalchromosome, followed at a postzygotic state by a non-reciprocal transfer of the mutant sequence from thematernal to the paternal chromosome by mitotic geneconversion. Subsequently, two genetically differentpopulations of cells developed, so that the tissue wasmosaic, containing both homozygous and heterozygousmutant cells.
Gene conversion was originally observed as the non-Mendelian segregation of the products of a single mei-osis in fungi. The first molecular evidence for a gene-conversion-like event in humans was detected in theglobin gene family and was described as an event thatoccurred during meiosis (Slightom et al. 1980). Sincethen, gene conversion has been reported in many otherclustered human gene families, such as gene clusters forimmunoglobulins, HLA antigens, red-green visual pig-ments, CYP21A/B, and PKD1/PKD1-related genes(Bentley and Rabbits 1983; Kuhner et al. 1991; Collieret al. 1993; Reyniers et al. 1995; Watnick et al. 1998). Inmost of these cases, the conversions involve interlocusexchange between gene and pseudogene or a related genelocated on the same chromosome. Gene conversion hasalso been implicated in von Willebrand disease, wherethe gene and pseudogene are on different chromosomes(Eikenboom et al. 1994). With the exception of the HLAsystem and of cancer, gene conversion involving allelicexchange has been observed rarely. Mitotic gene con-version is even rarer in human genetic disorders. In vivoreversion of inherited mutations to normal in some casesof Fanconi anemia have been ascribed to homologousmitotic gene conversion (reviewed in Hirschhorn 2003).Jonkman et al. (1997) have presented evidence for amitotic gene conversion in the COL17A1 gene in acompound heterozygous proband with autosomal
Table 3 Heterozygous SOX9 Y440X nonsense mutations in CDpatients
Casea Karyotype/gender
Mutation Survivaltime
Campomelia
1 46,XY/F TAC fi TAG 4 years Severe2 46,XX/F TAC fi TAG >11 years Severe3 46,XY/M TAC fi TAG 4 years Mild4 46,XX/F TAC fi TAA Abortion Severe5 46,XY/M TAC fi TAA >4 years Severe6 46,XY/M TAC fi TAG 10 months Severe7 46,XX/F TAC fi TAG 90 min Severe8 46,XY/F TAC fi TAA Abortion Severe9 46,XX/F TAC fi TAG >2 years None10 46,XX/F TAC fi TAA ? ?
aCase references: 1 Wagner et al. (1994), 2 Meyer et al. (1997),3 Hageman et al. (1997), 4–10 unpublished
Fig. 5 Proposed sequence of events for the generation of thehomozygous Y440X mutation in patient AG. As a first step, a denovo Y440X mutation in SOX9 occurred in the maternal germline,indicated by an asterisk, resulting in a zygote that was heterozygousfor the mutant maternal SOX9 allele and for a normal paternalSOX9 allele. In a second independent step, after several celldivisions (short arrows), a somatic gene conversion led toreplacement of the wild-type sequence on the paternal allele bythe mutant maternal sequence, resulting in homozygous mutantcells. These constituted about 80% of the leukocyte cell population,whereas about 20% were heterozygous mutant cells. The degree ofmosaicism in other tissues is not known
50

recessive epidermolysis bullosa in whom the phenotypewas changed from mutant to the wild-type, leading torevertant mosaicism (the parents were heterozygouscarriers). In contrast, a mitotic gene conversion eventchanged a de novo heterozygous mutation to a homo-zygous mutation in the present case, which has, to ourknowledge, not been observed previously. Homologousrecombination by mitotic gene conversion has beenshown to be an important pathway for DNA repair,which is strongly induced by DNA double-strand breaks(Kourilsky 1986; Wiese et al. 2002) in mammalian cells.The frequency of mitotic gene conversion is greatestwhen the template for homologous repair is a sisterchromatid (Johnson and Jasin 2001). It is tempting tospeculate that the mutation reported here, which in-volved exchange between homologous chromosomesrather than between sister-chromatids, might be theoutcome of DNA mismatch repair ‘‘going wrong’’ in thesense that it resulted not in back-mutation to wild-type,but in forward mutation to mutant.
The fact that patient AG survived for 3 months afterbirth, despite carrying a homozygous nonsense muta-tion in SOX9, can be explained in several ways. First, asshown here for DNA from peripheral blood, not all cellscarried the mutation in homozygous form, and thismosaic situation could also apply to other tissues. Thepercentage of homozygous mutant cells in these un-sampled tissues could be higher or lower than the valueof �80% obtained for leukocytes. Unfortunately, DNAsamples from tissue of particular relevance for CD, suchas cartilage or gonads, were not available. However,even if the cell population homozygous for the Y440Xallele was in the majority in these tissues, this might stillbe compatible with development until after birth. Thisassumption derives from circumstantial and experi-mental evidence that the Y440X allele is not a completeloss-of-function allele but rather constitutes a hypo-morphic allele.
Circumstantial evidence is provided by the ten casescarrying the mutation in heterozygous form. This groupof CD cases resemble the group of CD translocationcases who have two intact copies of SOX9 with onecopy downregulated by interruption of the cis-actingregulation of SOX9 (Pfeifer et al. 1999). As with thetranslocation cases, the Y440X heterozygous cases areon average less severely affected than the majority ofCD cases. Excluding the two abortion cases (cases 4 and9) and case 10 with no available information, four ofseven patients survived for more than 2 years (Table 3)as did the majority of the translocation patients,whereas such long-term survivors constitute only about10% of all CD cases (Mansour et al. 1995; Pfeifer et al.1999). Likewise, overt campomelia is presented by about90% of all CD patients but is found in only seven ofnine Y440X cases, the other two having mild or nobending of lower limbs, as is the case for about half ofthe translocation cases.
Experimental evidence for the Y440X allele consti-tuting a hypomorphic allele is provided by our cell
transfection studies, which have revealed residualtransactivation activity for the mutant protein onauthentic SOX9 target genes in the range of 5% to 22%.A similar study had shown that progressive truncationof the C-terminal transactivation domain results inprogressive loss of transactivation ability, with residualactivities of about 20% obtained with truncations atresidues 454 and 437 (McDowall et al. 1999). Likewise,our previous studies have shown residual transactivationpotential for residues 402–439 fused to a heterologousGAL4 DNA-binding domain of 25% compared withfull-length SOX9, when tested on a GAL4-dependentreporter plasmid (Meyer et al. 1997). Therefore, even if atissue consisted mainly of Y440X homozygous mutantcells, the residual activity of, on average, about 15%could still suffice to allow development to proceed in thistissue mostly as it would in a classical CD case hetero-zygous for a SOX9 null allele, in which all cells have50% wild-type activity, depending on the SOX9 dosagesensitivity of the tissue in question.
Another potential explanation for the similarity ofthe skeletal phenotype of the present case to the classicalSOX9 heterozygote is selection for the heterozygouscells in cartilage. Since Sox9 null cells are unable toundergo chondrogenesis (Bi et al. 1999), the Y440Xhomozygous mutant cells might have failed to beincorporated into cartilage. Such selection is well docu-mented in disorders such as X-linked agammaglobulin-emia (Fearon et al. 1987).
Apart from the homozygous Y440X mutation inpatient AG, the same nonsense mutation has been ob-served in ten independent CD cases in heterozygousform (Table 3). This recurrent mutation accounts forabout 8% of all coding region mutations and is the onlymutational hot spot in SOX9. What might be the reasonfor the high mutation rate at this codon? Inspection ofthe sequence shows that the mutable cytosine 1692 atthe end of codon 440 is followed by a G residue, con-stituting a CpG dinucleotide (Fig. 2a). This dinucleotideis a well-known hot spot for mutations in human geneticdisease because of methylation of the C residue, which,following deamination, results in a C fi T transition(Cooper and Youssoufian 1998). This mechanism wouldresult in substitution of a TAC by a TAT codon atposition 440, both encoding tyrosine, and constitute asilent mutation without phenotypic effects. In contrast,the Y440X nonsense mutations consist of transversionsfrom C to A or G. Interestingly, several studies haveshown that transversion rates are elevated at methylatedCpG dinucleotides. Whereas transitions at these sitesare elevated about 20-fold relative to transitions at othersites, transversions at CpG sites are elevated aboutfivefold to eightfold (Bottema et al. 1991). The mecha-nism for the occurrence of transitions at methylatedCpG sites is known, that for the occurrence of trans-versions at these sites is not well understood. The factthat nonsense mutations have not been described so farat any of the other eleven TAC tyrosine codons in theTA domain of SOX9, none of which is followed by a
51

codon starting with a G residue, supports the view thatthe Y440X hot spot is caused by a mutational mecha-nism that leads to an elevated transversion rate at CpGdinucleotides. Sequence context may also play a role asis suggested by the observation that at the two otherTAC codons that are followed by a G nucleotide,mutation to a stop codon has been detected only oncefor codon 84 (unpublished).
In summary, this is the first report of a patient withCD who has a homozygous SOX9 mutation. Themechanism underlying the mutation has been demon-strated to be a de novo mutation followed by mitoticgene conversion. The somatic mosaicism combined withthe homozygosity for a hypomorphic allele rather than aloss-of-function allele contributed to the postnatal sur-vival in the patient, in what would otherwise most likelyrepresent a lethal situation.
Acknowledgements We thank the parents of the patient for theirinterest and support, Maureen Bocian for providing informationabout the AG family, Victor Steimle for access to and help with theLightCycler, and Michael Wegner for the Neuro2A cell line. Weare grateful to Drs. Mary Ann Floyd, Ephrat Levy-Lahad, JoanPaterson, Elie Picard, Rosario Santos, Eva Seemanova, NielsTommerup, and William Wilcox for submitting samples of andproviding information on patients with heterozygous Y440Xmutations, and to Maureen Bocian and Jurgen Kohlhase forcomments on the manuscript. We appreciate the constructivecomments made by one of the reviewers. Francis Poulat, L.Bridgewater, and Benoit de Crombrugghe are acknowledged forproviding plasmids. This work was supported by a grant from theDeutsche Forschungsgemeinschaft to G.S. (Sche 194/15–1 and –2).
References
Akiyama H, Chaboissier MC, Martin JF, Schedl A, CrombruggheB de (2002) The transcription factor Sox9 has essential roles insuccessive steps of the chondrocyte differentiation pathway andis required for expression of Sox5 and Sox6. Genes Dev16:2813–2828
Arango NA, Lovell-Badge R, Behringer RR (1999) Targetedmutagenesis of the endogenous mouse Mis gene promoter: invivo definition of genetic pathways of vertebrate sexual devel-opment. Cell 99:409–419
Bentley DL, Rabbits TH (1983) Evolution of immunoglobulin Vgenes: evidence indicating that recently duplicated human Vjsequences have diverged by gene conversion. Cell 32:181–189
Bernard P, Tang P, Liu S, Dewing P, Harley VR, Vilain E (2003)Dimerization of SOX9 is required for chondrogenesis, but notfor sex determination. Hum Mol Genet 12:1755–1765
Bi W, Deng JM, Zhang Z, Behringer RB, Crombrugghe B de(1999) Sox9 is required for cartilage formation. Nat Genet22:85–89
Bottema CDK, Bottema MJ, Ketterling RP, Yoon HS, Janco RL,Phillips JA III, Sommer SS (1991) Why does the human factorIX gene have a G + C content of 40%? Am J Hum Genet49:839–850
Bridgewater LC, Lefebvre V, Crombrugghe B de (1998) Chon-drocyte-specific enhancer elements in the Col11a2 gene resemblethe Col2a1 tissue-specific enhancer. J Biol Chem 273:14998–15006
Bridgewater LC, Walker MD, Miller GC, Ellison TA, HolsingerLD, Potter JL, Jackson TL, Chen RK, Winkel VL, Zhang Z,McKinney S, Crombrugghe B de (2003) Adjacent DNA
sequences modulate Sox9 transcriptional activation at pairedSox sites in three chondrocyte-specific enhancer elements. Nu-cleic Acids Res 31:1541–1553
Cavenee WK, Dryja TP, Phillips RA, Benedict WF, Godbout R,Gallie BL, Murphree AL, Strong LC, White RL (1993)Expression of recessive alleles by chromosomal mechanisms inretinoblastoma. Nature 305:779–784
Chaboissier MC, Kobayashi A, Vidal VIP, Lutzkendorf S,Kant HJK van de, Wegner M, Rooij DG de, Behringer RR,Schedl A (2004) Functional analysis of Sox8 and Sox9during sex determination in the mouse. Development 131:1891–1901
Collier S, Tassabehji M, Sinnott P, Strachan T (1993) A de novopathological point mutation at the 21-hydroxylase locus:implications for gene conversion in the human genome. NatGenet 3:260–265
Cooper DN, Youssouffian H (1988) The CpG dinucleotide andhuman genetic disease. Hum Genet 78:151–155
Eikenboom JCJ, Vink T, Briet E, Sixma JJ, Reitsma PH (1994)Multiple substitutions in the von Willebrand factor gene thatmimic the pseudogene sequence. Proc Natl Acad Sci USA91:2221–2224
Fearon ER, Winkelstein JA, Civin CI, Pardoll DM, VogelsteinB(1987) Carrier detection in X-linked agammaglobulinemia byanalysis of X-chromosome inactivation. N Engl J Med 316:427–431
Foster JW, Dominguez-Steglich MA, Guioli S, Kwok C, WellerPA, Stevanovic M, Weissenbach J, Mansour S, Young ID,Goodfellow PN, Brook JD, Schafer AJ (1994) Campomelicdysplasia and autosomal sex reversal caused by mutations in anSRY-related gene. Nature 372:525–530
Hageman RM, Cameron FJ, Sinclair AH (1998) Mutation analysisof the SOX9 gene in a patient with campomelic dysplasia. HumMutat Suppl 1:S112–S113
Hirschhorn R (2003) In vivo reversion to normal of inheritedmutations in humans. J Med Genet 40:721–728
Houston CS, Opitz JM, Spranger JW, Macpherson RI, Reed MH,Gilbert EF, Herrmann J, Schinzel A (1983) The campomelicsyndrome: review, report of 17 cases, and follow-up on thecurrently 17-year-old boy first reported by Maroteaux et al. in1971. Am J Med Genet 15:3–28
Johnson RD, Jasin M (2001) Double-strand-break-inducedhomologous recombination in mammalian cells. Biochem SocTrans 29:196–201
Jonkman MF, Scheffer H, Stulp R, Pas HH, Nijenhuis M, HeeresK, Owaribe K, Pulkkinen L, Uitto J (1997) Revertant mosai-cism in epidermolysis bullosa caused by mitotic gene conver-sion. Cell 88:543–551
Kent J, Wheatley SC, Andrews JE, Sinclair AH, Koopman P(1996) A male-specific role for SOX9 in vertebrate sex deter-mination. Development 122:2813–2822
Kourilsky P (1986) Molecular mechanisms for gene conversion inhigher cells. Trends Genet 2:60–63
Kuhner MK, Lawlor DA, Ennis PD, Parham P (1991) Gene con-version in the evolution of the human and chimpanzee MHCclass I loci. Tissue Antigens 38:152–164
Lefebvre V, Huang W, Harley VR, Goodfellow PN, CrombruggheB de (1997) SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol CellBiol 17:2336–1246
Mansour S, Hall CM, Pembrey ME, Young ID (1995) A clinicaland genetic study of campomelic dysplasia. J Med Genet32:415–420
McDowall S, Argentaro A, Ranganathan S, Weller P, Mertin S,Mansour S, Tolmie J, Harley V (1999) Functional and struc-tural studies of wild type SOX9 and mutations causing cam-pomelic dysplasia. J Biol Chem 274:24023–24030
Meyer J, Sudbeck P, Held M, Wagner T, Schmitz ML, BricarelliFD, Eggermont E, Friedrich U, Haas OA, Kobelt A, LeroyJG, Maldergem L van, Michel E, Mitulla B, Pfeiffer RA,Schinzel A, Schmidt H, Scherer G (1997) Mutational analysis
52

of the SOX9 gene in campomelic dysplasia and autosomal sexreversal: lack of genotype/phenotype correlations. Hum MolGenet 6:91–98
Morais da Silva S, Hacker A, Harley V, Goodfellow P, Swain A,Lovell-Badge R (1996) Sox9 expression during gonadal devel-opment implies a conserved role for the gene in testis differen-tiation in mammals and birds. Nat Genet 14:62–68
Ng LJ, Wheatley S, Muscat GEO, Conway-Campbell J, Bowles J,Wright E, Bell DM, Tam PP, Cheah KS, Koopman P (1997)SOX9 binds DNA, activates transcription, and coexpresses withtype II collagen during chondrogenesis in the mouse. Dev Biol183:108–121
Olney PN, Kean LS, Graham D, Elsas LJ, May KM (1999)Campomelic syndrome and deletion of SOX9. Am J Med Genet84:20–24
Pfeifer D, Kist R, Dewar K, Devon K, Lander ES, Birren B,Korniszewski L, Back E, Scherer G (1999) Campomelic dys-plasia translocation breakpoints are scattered over 1 Mbproximal to SOX9: evidence for an extended control region.Am J Hum Genet 65:111–124
Pop R, Conz C, Lindenberg KS, Blesson S, Schmalenberger B,Briault S, Pfeifer D, Scherer G (2004) Screening of the 1 MbSOX9 5¢ control region by array CGH identifies a large deletionin a case of campomelic dysplasia with XY sex reversal. J MedGenet 41:e47
Reyniers E, Thienen MN van, Meire F, Boulle K de, Devries K,Kestelijn P, Willems PJ (1995) Gene conversion between redand defective green opsin gene in blue cone monochromacy.Genomics 29:323–328
Santa Barbara P de, Bonneaud N, Boizet B, Desclozeaux M,Moniot B, Sudbeck P, Scherer G, Poulat F, Berta P (1998)
Direct interaction of SRY-related protein SOX9 and steroido-genic factor 1 regulates transcription of the human anti-Mullerian hormone gene. Mol Cell Biol 18:6653–6665
Slightom JL, Blechl AE, Smithies O (1980) Human fetal Gc- andAc-globin genes: complete nucleotide sequences suggest thatDNA can be exchanged between these duplicate genes. Cell21:627–638
Sock E, Pagon RA, Keymolen K, Lissens W, Wegner M, Scherer G(2003) Loss of DNA-dependent dimerization of the transcrip-tion factor SOX9 as a cause for campomelic dysplasia. HumMol Genet 12:1439–1447
Sudbeck P, Schmitz ML, Baeuerle PA, Scherer G (1996) Sexreversal by loss of the C-terminal transactivation domain ofhuman SOX9. Nat Genet 13:230–232
Vidal VP, Chaboissier MC, Rooij DG de, Schedl A (2001) Sox9induces testis development in XX transgenic mice. Nat Genet28:216–217
Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, PasantesJ, Dagna Bricarelli F, Keutel J, Hustert E, Wolf U, TommerupN, Schempp W, Scherer G (1994) Autosomal sex reversal andcampomelic dysplasia are caused by mutations in and aroundthe SRY-related gene SOX9. Cell 79:1111–1120
Watnick TJ, Gandolph MA, Weber H, Neumann HPH, GerminoGG (1998) Gene conversion is a likely cause of mutation inPKD1. Hum Mol Genet 7:1239–1243
Wiese C, Pierce AJ, Gauny SS, Jasin M, Kronenberg A (2002)Gene conversion is strongly induced in human cells by double-strand breaks and is modulated by the expression of BCL-x(L).Cancer Res 62:1279–1283
53


















