
A DFT study in bulk magnetic moment of FexCo1–x (0 ≤ x ≤ 1)
4
A DFT study in bulk magnetic moment of Fe x Co 1–x (0 £ x £ 1) M SOLORZA-GUZMA ´ N 1 , G RAMI ´ REZ-DA ´ MASO 2, * and F L CASTILLO-ALVARADO 3 1 Escuela Superior de Co ´mputo del Instituto Polite ´cnico Nacional, C. P. 07738 Ciudad de Me ´xico, Me ´xico 2 Escuela Superior de Ingenierı ´a y Arquitectura ‘‘Unidad Ticoma ´n’’ del Instituto Polite ´cnico Nacional, C. P. 07340 Ciudad de Me ´xico, Me ´xico 3 Escuela Superior de Fı ´sica y Matema ´ticas del Instituto Polite ´cnico Nacional, C. P. 07738 Ciudad de Me ´xico, Me ´xico *Author for correspondence ([email protected]) MS received 2 November 2020; accepted 28 December 2020 Abstract. In this study, we present results of the electronic density of states (DOS) and bulk magnetic moment of iron (Fe), cobalt (Co) and their alloys (Fe x Co 1–x ; x = 1.0, 0.95, …, 0.0). Density functional theory with the generalized gradient approximation was applied to obtain geometric and electronic properties. The methodology uses virtual crystal approxi- mation, in conjunction with CASTEP module and the functionals PBE and PBESol of the molecular simulation program Material Studio. We optimized the geometry of the bulk (obtaining their lattice parameters), which the structure was used to determine the bulk magnetic moments. To determine the magnetic moment, we calculated the difference of the electronic DOS of the electrons with spin up and spin down. The geometric optimization and magnetic moment obtained in the present study are very similar to the experimental results, with a maximum error of 8%, which makes the present article interesting. Keywords. Iron–cobalt alloy; bulk magnetic moment; electronic density of states. 1. Introduction It is very well known that iron (Fe), cobalt (Co) and their alloys have one of the major magnetic moments, with an experimental value of 2.22 l B for Fe, 1.72 l B for Co [1] and 2.42 l B for Fe 0.5 Co 0.5 [2]. The magnetic properties of Fe 0.5 Co 0.5 have been studied by different authors experi- mentally [2–4] or theoretically [5–9]. The study of properties of iron–cobalt equiatomic has been done in bulk [2,4,6] as a homogenous material, or over the surface [7–9], with the inhomogeneity or discontinuity of the surface. Bardos [2] reported the influence of composition and the temperature rate quenching of mean magnetic moment of iron–cobalt alloy. Asano et al [3] reported the specific heat and the lattice parameter of ordered and disordered FeCo alloy. Also, dif- ferent authors have reported the value of magnetic moment on the surface, with an increase, about bulk of the order of 30%. This increase in the surface magnetic moment compared with bulk magnetic moment is mainly due to the nearest number neighbour atoms in bulk and on the surface. In previous works [8,9], we used a phenomenological method, where we studied magnetization as a function of temperature and aspects related to the surface, because thin films were considered. In this article, we present a methodology to compute their den- sity of states and magnetic moment of Fe x Co 1–x binary alloy with a quantum mechanical method. Magnetic materials represent the core of expanding business, many industrial electronic components are based on nanocrystalline thin-film media and magneto-optic materials that were not available 20 years ago. At the beginning of this century, O’Handley and Robert [10] have described a more efficient and punctual method in the manufacture of electronic materials and components, as well as electrochemical and electromagnetic systems, where Fe x Co 1–x alloys were used. The magnetism (in bulk or on the surface) of disordered binary alloys has been exten- sively and experimentally studied, the different theoretical models that have tried to explain the observed results are based on crude approximations, such as the Bethe’s lattice. The experimental part has shown that the magnetic prop- erties on the surface of iron are different from those in the bulk, while in the theory of magnetism it is not possible to predict these variations in magnetic properties between bulk and surface. The methods of approximation developed by ab initio theory can be divided in two different groups. The first one is based on the wavefunction [11]. For example, when we study disordered binary alloys, it is always accompanied with other methods, concepts and assumptions, with very large clusters of atoms as the clusters–Bethe–lattice method (CBLM) with a strong link Hamiltonian, to calculate the internal energy of the system, and combining it with the cluster variation method proposed by Kikuchi [12], which give us the configurational entropy of the alloy. However in general, the disadvantage of this method is that their application to non-homogeneous systems such as atoms, Bull. Mater. Sci. (2021) 44:93 Ó Indian Academy of Sciences https://doi.org/10.1007/s12034-021-02412-7
Transcript of A DFT study in bulk magnetic moment of FexCo1–x (0 ≤ x ≤ 1)

A DFT study in bulk magnetic moment of FexCo1–x (0 £ x £ 1)
M SOLORZA-GUZMAN1, G RAMIREZ-DAMASO2,* and F L CASTILLO-ALVARADO3
1 Escuela Superior de Computo del Instituto Politecnico Nacional, C. P. 07738 Ciudad de Mexico, Mexico2 Escuela Superior de Ingenierıa y Arquitectura ‘‘Unidad Ticoman’’ del Instituto Politecnico Nacional,
C. P. 07340 Ciudad de Mexico, Mexico3 Escuela Superior de Fısica y Matematicas del Instituto Politecnico Nacional, C. P. 07738 Ciudad de Mexico, Mexico
*Author for correspondence ([email protected])
MS received 2 November 2020; accepted 28 December 2020
Abstract. In this study, we present results of the electronic density of states (DOS) and bulk magnetic moment of iron (Fe),
cobalt (Co) and their alloys (FexCo1–x; x = 1.0, 0.95, …, 0.0). Density functional theory with the generalized gradient
approximation was applied to obtain geometric and electronic properties. The methodology uses virtual crystal approxi-
mation, in conjunction with CASTEP module and the functionals PBE and PBESol of the molecular simulation program
Material Studio. We optimized the geometry of the bulk (obtaining their lattice parameters), which the structure was used to
determine the bulk magnetic moments. To determine the magnetic moment, we calculated the difference of the electronic
DOS of the electrons with spin up and spin down. The geometric optimization and magnetic moment obtained in the present
study are very similar to the experimental results, with a maximum error of 8%, which makes the present article interesting.
Keywords. Iron–cobalt alloy; bulk magnetic moment; electronic density of states.
1. Introduction
It is very well known that iron (Fe), cobalt (Co) and their
alloys have one of the major magnetic moments, with an
experimental value of 2.22 lB for Fe, 1.72 lB for Co [1] and
2.42 lB for Fe0.5Co0.5 [2]. The magnetic properties of
Fe0.5Co0.5 have been studied by different authors experi-
mentally [2–4] or theoretically [5–9]. The study of properties
of iron–cobalt equiatomic has been done in bulk [2,4,6] as a
homogenous material, or over the surface [7–9], with the
inhomogeneity or discontinuity of the surface. Bardos [2]
reported the influence of composition and the temperature
rate quenching of mean magnetic moment of iron–cobalt
alloy. Asano et al [3] reported the specific heat and the lattice
parameter of ordered and disordered FeCo alloy. Also, dif-
ferent authors have reported the value of magnetic moment on
the surface, with an increase, about bulk of the order of 30%.
This increase in the surface magnetic moment compared with
bulk magnetic moment is mainly due to the nearest number
neighbour atoms in bulk and on the surface. In previous works
[8,9], we used a phenomenological method, where we studied
magnetization as a function of temperature and aspects
related to the surface, because thin films were considered. In
this article, we present a methodology to compute their den-
sity of states and magnetic moment of FexCo1–x binary alloy
with a quantum mechanical method.
Magnetic materials represent the core of expanding
business, many industrial electronic components are based
on nanocrystalline thin-film media and magneto-optic
materials that were not available 20 years ago. At the
beginning of this century, O’Handley and Robert [10] have
described a more efficient and punctual method in the
manufacture of electronic materials and components, as
well as electrochemical and electromagnetic systems, where
FexCo1–x alloys were used. The magnetism (in bulk or on
the surface) of disordered binary alloys has been exten-
sively and experimentally studied, the different theoretical
models that have tried to explain the observed results are
based on crude approximations, such as the Bethe’s lattice.
The experimental part has shown that the magnetic prop-
erties on the surface of iron are different from those in the
bulk, while in the theory of magnetism it is not possible to
predict these variations in magnetic properties between bulk
and surface.
The methods of approximation developed by ab initiotheory can be divided in two different groups. The first one
is based on the wavefunction [11]. For example, when we
study disordered binary alloys, it is always accompanied
with other methods, concepts and assumptions, with very
large clusters of atoms as the clusters–Bethe–lattice method
(CBLM) with a strong link Hamiltonian, to calculate the
internal energy of the system, and combining it with the
cluster variation method proposed by Kikuchi [12], which
give us the configurational entropy of the alloy. However in
general, the disadvantage of this method is that their
application to non-homogeneous systems such as atoms,
Bull. Mater. Sci. (2021) 44:93 � Indian Academy of Scienceshttps://doi.org/10.1007/s12034-021-02412-7Sadhana(0123456789().,-volV)FT3](0123456789().,-volV)

molecules, solids or surfaces has a high computational cost.
The second method is based on the density functional the-
ory (DFT) [13–15], which studies the properties of each one
of the nodes with electronic density of charge qelec r~ð Þ,avoiding the explicit determination of the wavefunction
uelec; that is, uses an approximation of the Hamiltonian and
obtains an accurate density of charge, including the
dynamic correlation due to the Coulomb interactions
between electrons. This approximation presents a unique
problem: the exchange-correlation functional and potential
derivative therein, the solution requires a good approxi-
mation to the exchange-correlation functional. The most
relevant functionals are local density approximation (LDA),
the generalized gradient approximation (GGA), the
metaGGA and hybrid functionals. All these functionals are
very efficient and accurate; however, the results obtained
for metals are more accurate with the GGA [16,17].
In the present study, compositional disorder was con-
sidered as the probability of finding an atom of Fe or Co in a
site at the centre or at the corner of a bcc crystal structure.
Electronic density of states (DOS) is obtained after that bulk
FeCo was optimized, using different approximation meth-
ods developed in this program. We select the best func-
tionals of GGA as PBE and PBESol, to determine the DOS
and the magnetic moment that had better approximate to
experimental value.
2. Methodology and computational details
Virtual crystal approximation (VCA) is a technique that
considers the periodicity of the primitive cell and consider
that on each site, there is a virtual atom that interpolates
between the behaviours of both atoms. On the other hand,
the ab initio method is generally well suited for the
approximation (VCA) because it is possible to solve prob-
lems more accurately and molecules with basal non-
degenerate states. The importance in the Fe0.5Co0.5 alloy is
the difference between the kinetic energy of a node of
configurational structure with electrons of independent
valences and the first interacting neighbours due to Cou-
lomb interaction between electrons. This dynamic correla-
tion is crucial in DFT theory, which allows us an accurate
description of the interaction of many-particle systems in
terms of an effective system of non-interacting particles
with a density functional, whose formulism is not complete
with the exchange-correlation functional; requires a good
approximation in terms of this interaction correlation.
To treat this functional exchange-correlation energy
(EXC) in terms of the DOS, it is coupled with the functional
improved with GGA approximation and the computer pro-
gram developed in CASTEP of Material Studio [18–20];
this program uses the method of a pseudopotential plane
wave, allowing to develop calculations in quantum
mechanics first-principles.
2.1 Geometry optimization of bulk FexCo1–x
The first step is to construct a crystal structure of Fe (bcc) or
Co (fcc) included in the library of Material Studio program.
For FexCo1–x alloys (x = 1.00, 0.95, …, 0.30), we use a bcc
unit cell with the mentioned concentrations and for x = 0.25,
0.20, ..., 0.00, we use a fcc unit cell. Body centred cubic
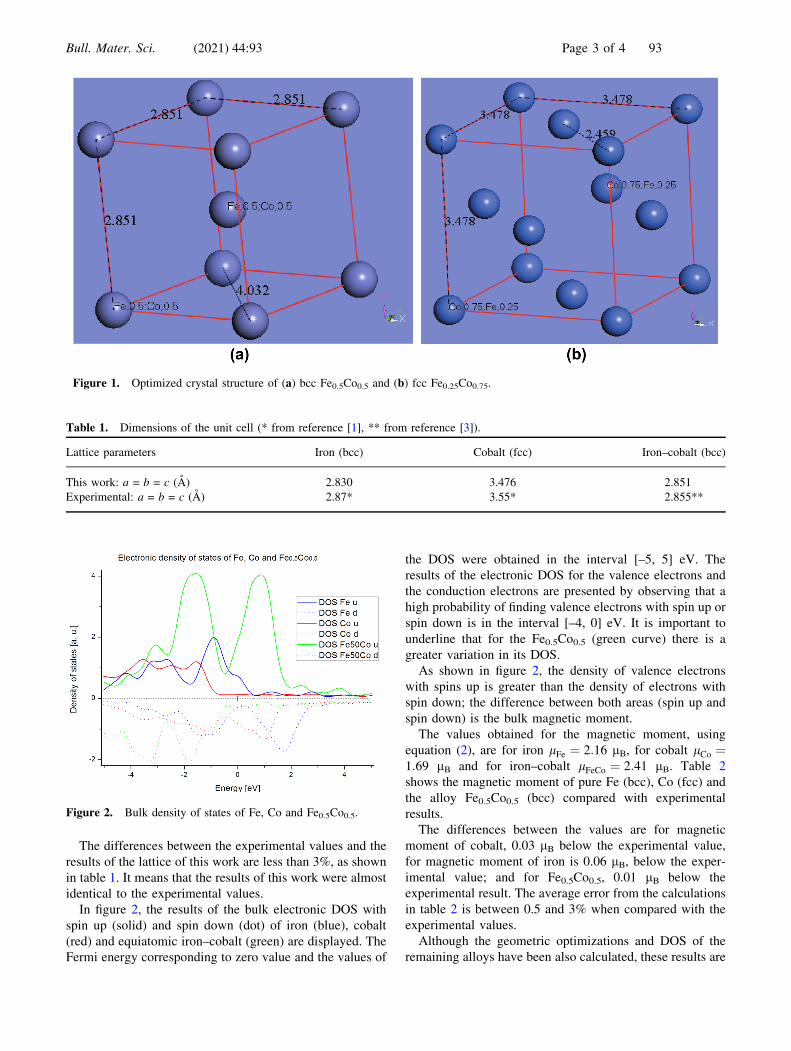
Fe0.5Co0.5 crystal structures are shown in figure 1a and face
centred cubic Fe0.25Co0.75 unit cell is shown in figure 1b.
The optimized crystal structure gives us the state of
minimum energy and the electronic DOS of the alloy.
2.2 DOS and magnetic moment
The electronic DOS is a function that allows knowing the
properties of materials. For instance, knowing whether it is
a conductor, insulator or semiconductor. In this work, DOS
allows determining the magnetic moment of the FexCo1–x
alloys.
The spin up with a" of atom a (Fe, Co) and spin down b#of atom b (Fe, Co) were designated, with a cutoff energy of
300 eV of the basic set of plane waves. The density of
points in the Brillouin zone was considered with k-point
grid, corresponding to a 3 � 4 � 1 Monkhorst-Pack grid.
The convergence of the geometric optimization is carried
out with the following values: 5 9 10-6 eV per atom for
maximum energy change, 0.01 eV A-1 as the maximum
force, 0.02 GPa as the maximum stress and 5 9 10-4 A for
the maximum displacement tolerance. In this part, the
functionals PBE and PBESol were considered to obtain the
most accurate results of bulk magnetic moments.
Using the procedure outlined above, the DOS for elec-
trons with spin up qa"� �
of atoms a Fe, Coð Þ and the
electronic DOS with spin down qb#� �
of atoms b Fe, Coð Þare obtained. Then, the average number of electrons with
spin up hn"i and the number of electrons with spin down
hn#i are calculated by:
hndi ¼Z
qd eð Þde; with d ¼"; # ð1Þ
Finally, magnetic moments are calculated as:
l ¼ hn"i � hn#i ð2Þ
in units of Bohr magneton.
3. Results and discussion
As mentioned previously, the first step in this work was to
optimize the geometry of the unit cell. In this process, the
optimal parameters of the crystal lattice are obtained. In
table 1, experimental results compared with our results of
the optimized unit cell of iron, cobalt and iron–cobalt alloy
are presented.
93 Page 2 of 4 Bull. Mater. Sci. (2021) 44:93
The differences between the experimental values and the
results of the lattice of this work are less than 3%, as shown
in table 1. It means that the results of this work were almost
identical to the experimental values.
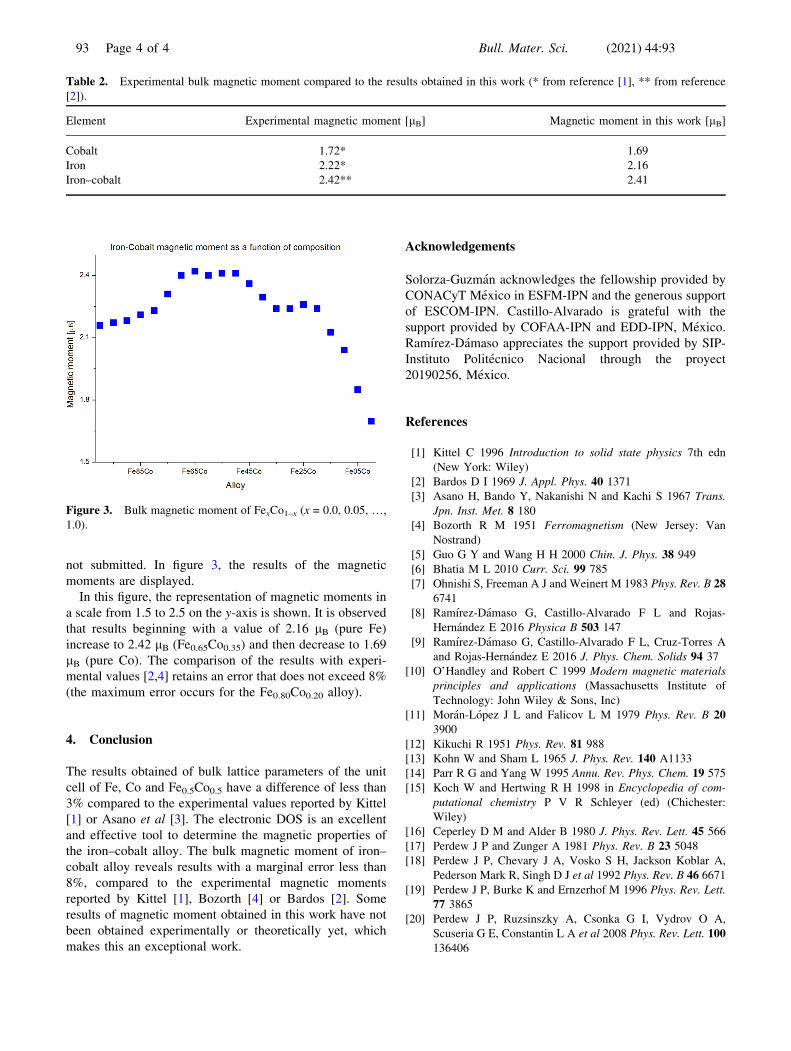
In figure 2, the results of the bulk electronic DOS with
spin up (solid) and spin down (dot) of iron (blue), cobalt
(red) and equiatomic iron–cobalt (green) are displayed. The
Fermi energy corresponding to zero value and the values of
the DOS were obtained in the interval [–5, 5] eV. The
results of the electronic DOS for the valence electrons and
the conduction electrons are presented by observing that a
high probability of finding valence electrons with spin up or
spin down is in the interval [–4, 0] eV. It is important to
underline that for the Fe0.5Co0.5 (green curve) there is a
greater variation in its DOS.
As shown in figure 2, the density of valence electrons
with spins up is greater than the density of electrons with
spin down; the difference between both areas (spin up and
spin down) is the bulk magnetic moment.
The values obtained for the magnetic moment, using
equation (2), are for iron lFe ¼ 2:16 lB, for cobalt lCo ¼1:69 lB and for iron–cobalt lFeCo ¼ 2:41 lB. Table 2
shows the magnetic moment of pure Fe (bcc), Co (fcc) and
the alloy Fe0.5Co0.5 (bcc) compared with experimental
results.
The differences between the values are for magnetic
moment of cobalt, 0.03 lB below the experimental value,
for magnetic moment of iron is 0.06 lB, below the exper-
imental value; and for Fe0.5Co0.5, 0.01 lB below the
experimental result. The average error from the calculations
in table 2 is between 0.5 and 3% when compared with the
experimental values.
Although the geometric optimizations and DOS of the
remaining alloys have been also calculated, these results are
Figure 1. Optimized crystal structure of (a) bcc Fe0.5Co0.5 and (b) fcc Fe0.25Co0.75.
Table 1. Dimensions of the unit cell (* from reference [1], ** from reference [3]).
Lattice parameters Iron (bcc) Cobalt (fcc) Iron–cobalt (bcc)
This work: a = b = c (A) 2.830 3.476 2.851
Experimental: a = b = c (A) 2.87* 3.55* 2.855**
Figure 2. Bulk density of states of Fe, Co and Fe0.5Co0.5.
Bull. Mater. Sci. (2021) 44:93 Page 3 of 4 93
not submitted. In figure 3, the results of the magnetic
moments are displayed.
In this figure, the representation of magnetic moments in
a scale from 1.5 to 2.5 on the y-axis is shown. It is observed
that results beginning with a value of 2.16 lB (pure Fe)
increase to 2.42 lB (Fe0.65Co0.35) and then decrease to 1.69
lB (pure Co). The comparison of the results with experi-
mental values [2,4] retains an error that does not exceed 8%
(the maximum error occurs for the Fe0.80Co0.20 alloy).
4. Conclusion
The results obtained of bulk lattice parameters of the unit
cell of Fe, Co and Fe0.5Co0.5 have a difference of less than
3% compared to the experimental values reported by Kittel
[1] or Asano et al [3]. The electronic DOS is an excellent
and effective tool to determine the magnetic properties of
the iron–cobalt alloy. The bulk magnetic moment of iron–
cobalt alloy reveals results with a marginal error less than
8%, compared to the experimental magnetic moments
reported by Kittel [1], Bozorth [4] or Bardos [2]. Some
results of magnetic moment obtained in this work have not
been obtained experimentally or theoretically yet, which
makes this an exceptional work.
Acknowledgements
Solorza-Guzman acknowledges the fellowship provided by
CONACyT Mexico in ESFM-IPN and the generous support
of ESCOM-IPN. Castillo-Alvarado is grateful with the
support provided by COFAA-IPN and EDD-IPN, Mexico.
Ramırez-Damaso appreciates the support provided by SIP-
Instituto Politecnico Nacional through the proyect
20190256, Mexico.
References
[1] Kittel C 1996 Introduction to solid state physics 7th edn
(New York: Wiley)
[2] Bardos D I 1969 J. Appl. Phys. 40 1371
[3] Asano H, Bando Y, Nakanishi N and Kachi S 1967 Trans.Jpn. Inst. Met. 8 180
[4] Bozorth R M 1951 Ferromagnetism (New Jersey: Van
Nostrand)
[5] Guo G Y and Wang H H 2000 Chin. J. Phys. 38 949
[6] Bhatia M L 2010 Curr. Sci. 99 785
[7] Ohnishi S, Freeman A J and Weinert M 1983 Phys. Rev. B 286741
[8] Ramırez-Damaso G, Castillo-Alvarado F L and Rojas-
Hernandez E 2016 Physica B 503 147
[9] Ramırez-Damaso G, Castillo-Alvarado F L, Cruz-Torres A
and Rojas-Hernandez E 2016 J. Phys. Chem. Solids 94 37
[10] O’Handley and Robert C 1999 Modern magnetic materialsprinciples and applications (Massachusetts Institute of
Technology: John Wiley & Sons, Inc)
[11] Moran-Lopez J L and Falicov L M 1979 Phys. Rev. B 203900
[12] Kikuchi R 1951 Phys. Rev. 81 988
[13] Kohn W and Sham L 1965 J. Phys. Rev. 140 A1133
[14] Parr R G and Yang W 1995 Annu. Rev. Phys. Chem. 19 575
[15] Koch W and Hertwing R H 1998 in Encyclopedia of com-putational chemistry P V R Schleyer (ed) (Chichester:
Wiley)
[16] Ceperley D M and Alder B 1980 J. Phys. Rev. Lett. 45 566
[17] Perdew J P and Zunger A 1981 Phys. Rev. B 23 5048
[18] Perdew J P, Chevary J A, Vosko S H, Jackson Koblar A,
Pederson Mark R, Singh D J et al 1992 Phys. Rev. B 46 6671
[19] Perdew J P, Burke K and Ernzerhof M 1996 Phys. Rev. Lett.77 3865
[20] Perdew J P, Ruzsinszky A, Csonka G I, Vydrov O A,
Scuseria G E, Constantin L A et al 2008 Phys. Rev. Lett. 100136406
Table 2. Experimental bulk magnetic moment compared to the results obtained in this work (* from reference [1], ** from reference
[2]).
Element Experimental magnetic moment [lB] Magnetic moment in this work [lB]
Cobalt 1.72* 1.69
Iron 2.22* 2.16
Iron–cobalt 2.42** 2.41
Figure 3. Bulk magnetic moment of FexCo1–x (x = 0.0, 0.05, …,
1.0).
93 Page 4 of 4 Bull. Mater. Sci. (2021) 44:93
![Ê5MS=-0bandi.chungbuk.ac.kr/~ysk/phy4_2012.pdf · 2011-05-19 · - 83 - ÊU(x)≡[Ei(bulk)-Ei(x)]/q ÊUs≡[Ei(bulk)-Ei(0)]/q ÊUF≡[Ei(bulk)-EF]/q kT q ln(NA/ni) for p-type kT](https://static.fdocuments.net/doc/165x107/5ea6b41bb11a5121f72e6e2d/5ms-yskphy42012pdf-2011-05-19-83-uxaeibulk-eixq-usaeibulk-ei0q.jpg)












![Introduction to DFT+U - pitp.physics.ubc.ca · Introduction to DFT+U, Michel Côté, ... X ii 0 a † i t ii0 a i0 + 1 2 X ii jj ... The new functional is: ELDA+U[n]=ELDA[n]+EU [n](https://static.fdocuments.net/doc/165x107/5af86cea7f8b9a5f588cc0b4/introduction-to-dftu-pitp-to-dftu-michel-ct-x-ii-0-a-i-t-ii0-a-i0.jpg)


![kn DFT [x n )] =x n e = x(n e = x( N k - vvcet.ac.in · 16.How many multiplications and additions are required to compute N-point DFT using radix-2 FFT? In computing N-point DFT by](https://static.fdocuments.net/doc/165x107/5e0ff2adf1dabe726a5dbcdd/kn-dft-x-n-x-n-e-xn-e-x-n-k-vvcetacin-16how-many-multiplications.jpg)

