
3.2 Spettrometria MALDI-ToF
14
UNIVERSITÀ DEGLI STUDI DI MILANO FACOLTÀ DI MEDICINA VETERINARIA DIPARTIMENTO DI PATOLOGIA ANIMALE, IGIENE E SANITÀ PUBBLICA VETERINARIA – SEZIONE DI BIOCHIMICA E FISIOLOGIA VETERINARIA Materiale integrativo on-line al corso di TECNICHE DI PROTEOMICA Docente: Prof.ssa Gabriella Tedeschi
description
spettrometria document
Transcript of 3.2 Spettrometria MALDI-ToF

UNIVERSITÀ DEGLI STUDI DI MILANO FACOLTÀ DI MEDICINA VETERINARIA
DIPARTIMENTO DI PATOLOGIA ANIMALE,
IGIENE E SANITÀ PUBBLICA VETERINARIA – SEZIONE DI BIOCHIMICA E FISIOLOGIA
VETERINARIA
Materiale integrativo on-line al corso di TECNICHE DI PROTEOMICA
Docente: Prof.ssa Gabriella Tedeschi

3.2 SPETTROMETRIA DI MASSA MALDI-TOF La spettrometria di massa permette la identificazione delle sostanze attraverso la determinazione molto accurata della massa molecolare delle molecole intere o di loro frammenti opportunamente generati. Nel corso di tale analisi si possono distinguere due processi principali:
- generazione degli “ioni” (nella SORGENTE) - analisi del rapporto massa/carica” (m/z) degli ioni stessi
(nell’ANALIZZATORE) Tali processi possono aver luogo secondo modalità differenti dipendentemente del tipo di spettrometro di massa utilizzato. Nel caso particolare di studi di proteomica, il tipo di spettrometria di massa più diffuso e versatile è basato sulle tecnica MALDI-TOF (Matrix Assisted Laser Desorption/Ionization – Time Of Flight), un tipo di spettrometria di massa che permette l’analisi di peptidi e proteine con peso molecolare da poche centinaia a centinaia di migliaia di Da. Il nome di tale tecnica deriva appunto dal fatto che In questi spettrometri il processo di generazione degli ioni è ottenuto mediante la tecnica MALDI, mentre l’analisi del rapporto m/z è ottenuto mediante la tecnica TOF.
Generazione degli ioni
La tecnica MALDI consente di volatilizzare/ionizzare macromolecole grazie “all’assistenza” della matrice, ossia di molecole organiche (solitamente acidi aromatici) che vengono co-cristallizzate col campione di interesse (cositituito da peptidi o proteine pure o in miscela) su un opportuno supporto metallico (Target Plate, vedi Fig. 2). Il plate è posto nella “SORGENTE” (ossia in una camera ad alto vuoto, < 10 –7 torr) ed irradiato con impulsi laser della durata compresa tra 1 e 200 ns. Normalmente ci si avvale di un laser UV (337 nm) ad N2. La luce viene focalizzata tramite sistemi ottici singoli o multipli e passa attraverso una finestra nello spettrofotometro di massa. La posizione del fuoco del laser sul campione può essere cambiata sia muovendo l’asse ottico del sistema di focalizzazione sia muovendo il campione lungo degli assi fissi. Un parametro critico è l’irradiazione della luce laser sul campione. I migliori risultati si ottengono con una irradiazione che vada poco oltre la soglia di intensità utile per far volare la matrice.. Le molecole della matrice, assorbendo la radiazione laser, trasferiscono parte dell’energia assorbita all’analita, determinandone la volatilizzazione/ionizzazione. Il meccanismo con il quale si ottiene la volatilizzazione/ionizzazione del campione è analogo per tutte le molecole comprese nell’intervallo di applicabilità, anche se avviene con efficienza diversa in funzione sia del peso molecolare (l’efficienza diminuisce all’aumentare del peso molecolare), sia della natura del peptide o polipeptide (ossia, a parità di peso molecolare la volatilità di peptidi e proteine diverse è differente). In ogni caso, durante il processo di volatilizzazione/ionizzazione non si ha frammentazione dei campioni. Di conseguenza, si ottiene il peso molecolare dello “ione molecolare” (tanti quanti sono i componenti presenti nel campione), ossia della molecola+1 protone, da cui la simbologia [M+H]+. Gli ioni positivi formatisi vengono accellerati da un opportuno potenziale elettrico ed entrano nell’analizzatore (vedi paragrafo successivo). La Figura 2 descrive i processi che hanno luogo nella sorgente

Figura 2
Analisi del rapporto massa/carica (m/z) degli ioni
Alla metodica appena descritta è abbinata la tecnica TOF (Time Of Flight, “tempo di volo”) che permette la determinazione del rapporto massa/carica degli ioni generati dalla MALDI. In uno spettrofotometro TOF, un gruppo di ioni viene accelerato mediante un potenziale elettrico (generalmente compreso tra 1 e 30 kV -le esatte condizioni sperimentali per l’analisi TOF variano a seconda del campo di peso molecolare indagato e non saranno qui riportate-), che gli trasmette una determinata energia cinetica. La velocità degli ioni sarà proporzionale a (m/z)1/2, dove m/z è il rapporto massa/carica di un particolare tipo di ione. Gli ioni vengono poi fatti passare attraverso una regione libera da campi elettrici (di solito lunga da 0.1 a 3 m, chiamata DRIFT REGION o TUBO DI VOLO) dove si separano in una serie di gruppi di ioni identici, ognuno dei quali viaggia ad una velocità caratteristica della

propria massa. Alla fine di questa regione un rivelatore produce dei segnali ogni volta che un gruppo di ioni lo colpisce; la registrazione di questi segnali in funzione del tempo è uno spettro di massa TOF. In pratica, come descritto in dettaglio in un paragrafo successivo, il TEMPO DI VOLO, ossia il tempo impiegato tra il punto di partenza ed il punto di arrivo (comuni a tutti gli ioni) è proporzionale a (m/z)1/2 di ciascuno degli ioni stessi e può, quindi, essere usato per calcolare la massa degli ioni. L’analisi del tempo di volo può essere condotta mediante due modalità: LINEAR e REFLECTRON (Figure 3 e 4).
Figura 3: Analisi in modalità LINEAR. Uacc = potenziale di accellerazione; DRIFT REGION = tubo di volo
ANALIZZATORE SORGENTE ANALIZZATORE

Figura 4: Analisi in modalità REFLECTRON In pratica, in entrambe le modalità, gli ioni formati nella sorgente, che hanno tutti la stessa carica (z = +1), ma differenti masse, e quindi differenti (m/z), sono accelerati con velocità diverse e, dopo avere attraversato la regione di volo (DRIFT REGION) colpiscono il detector in tempi diversi. Agli ioni di una determinata massa è normalmente associato un certo range di energia, si hanno cioè ioni della stessa massa con velocità di accelerazione non perfettamente identiche, ma distribuite in un certo intervallo di valori. Tanto più è allargato tale intervallo, tanto più si hanno picchi segnale allargati e poco risolti.
SORGENTE ANALIZZATORE

Utilizzando un reflectron (che in pratica è assimilabile ad uno “specchio” di potenziali elettrici) è possibile compensare l’influenza della distribuzione di energia sul tempo di volo. Si ottiene così un dato migliore e picchi molto più stretti e risolti (fino ad un limite massimo di m/z di circa 5000).
Gli strumenti attualmente sono in grado di operare sia in modalità “linear” (regione di volo uguale a 1.5 m) per l’analisi di pesi molecolari ≥ 5000 Da circa ed in modalità “reflectron” (regione di volo uguale a 3 m) per analisi ottimale di masse ≤ 5000 Da.

Che cos’è uno spettro di massa MALDI-TOF? Il segnale rilevato dal rivelatore in seguito all’impatto degli ioni alla fine del tubo di volo è inviato ad un computer che lo elabora, trasformandolo in uno spettro di massa, ossia in un grafico i cui assi sono rappresentati da: -ascisse: m/z. Si tratta di una elaborazione ottenuta matematicamente in seguito a due processi: (1) trasformazione del tempo di volo in “m/z” e (2) calibrazione dello strumento, secondo quanto descritto di seguito -ordinate: intensità del segnale corrispondente ad un certo m/z (ossia quanti ioni di un determinato m/z hanno colpito il rivelatore). In realtà tale valore è solo molto approssimativamente correlato alla quantità dell’analita di quella massa presente nel campione, in quanto l’efficienza di volo di una certa specie dipende da molti fattori. Poichè la ionizzazione risultante dalla tecnica MALDI è condotta utilizzando condizioni che non provocano la frammentazione delle molecole proteiche; la conseguenza è che gli spettri di massa ottenuti sono di semplice interpretazione. Infatti per ogni specie presente nel campione si ottiene solitamente, un picco principale corrispondente alla massa della specie stessa di interesse, con l’aggiunta di un protone e con una singola carica elettrica (M + H)+ (per cui essendo z= +1, m/z = m) ed eventualmente un picco di dimensioni più ridotte corrispondente alla specie più due protoni, quindi con una doppia carica (M + H)2+, il cui m/z è pari ad ½ m/z del picco (M + H)+. In alcuni casi sono presenti anche picchi di intensità molto inferiore rispetto a (M + H)+ corrispondenti, ad esempio, a dimeri e trimeri del peptide [(2M + H)+ e (3M + H)+]. La valutazione del peso molecolare mediante calibrazione esterna prevede prima di tutto la calibrazione, ottenuta analizzando una miscela standard preparata (ed analizzata) contemporaneamente ai campioni ignoti. Principi teorici della trasformazione del tempo di volo in m/z e calibrazione: Le misure MALDI-TOF generano dati sotto forma di intensità di segnale ionico correlato al tempo di volo. Pertanto, per calcolare il dato m/z normalmente adottato nella spettrometria di massa è necessaria una trasformazione matematica del dato originario (che ha dimensioni di tempo). Questa trasformazione è ottenuta per mezzo di una procedura di calibrazione che usa proteine standard, ossia di massa nota. Prendendo in considerazione i principi della misura TOF si può ricavare l’equazione appropriata tramite la quale si possono determinare i coefficienti di calibrazione. Uno ione di massa m, carica z e dotato di una velocità v, ha bisogno di un certo tempo t per percorrere la distanza fissa s del tubo di volo fino al rivelatore. La velocità di uno ione (con carica elementare = e) accelerato mediante un campo elettrico determinato da una tensione U è data dall’equazione: v = ( 2 z e U/m )1/2 (1) dall’equazione (1), il tempo di volo di uno ione puo’ essere determinato come segue: t = s/v = ( s2/2 e U )1/2 ( m/z ) 1/2 (2)

Risistemando la (2) si puo’ ottenere la seguente relazione esistente tra m/z ed il tempo t: ( m/z )1/2 = ( 2 e U/s2 ) 1/2 t (3) Se il tempo zero è determinato dal segnale di sgancio (avviato mediante l’impulso laser) e se gli ioni iniziano a volare nello stesso istante, si trova la seguente relazione: ( m/z )1/2 = a1t (4) Il procedimento di calibrazione, quindi, permette di definire il coefficiente a1 mediante l’impiego dei valori TOF misurati per almeno due componenti (punti dicalibrazione) con valori di m/z noti e differenti tra loro. Ciò è ottenuto durante il processo di ” calibrazione” che può essere “esterno” o, più raramente, “interno”. La calibrazione esterna è ottenuta analizzando una miscela di sostanze di massa molecolare nota e simile a quella ipotizzabile per i(l) campioni(e) di interesse, utilizzando le medesime condizioni (voltaggio, modalità ecc.) che verranno poi utilizzate per l’analisi del campione. Le miscele “standard” variano quindi a seconda del tipo di campione da analizzare. Di seguito sono riportate le miscele (ed i relativi spettri di massa) utilizzate nel caso di analisi di peptidi di massa molecolare compresa tra circa 800 e circa 4000 Da e nel caso di proteine di massa molecolare compresa tra circa 20.000 e circa 80.000 Da. La calibrazione interna, usata più raramente, prevede che nel campione analizzato ci siano, oltre a sostanze ignote, almeno due componenti di cui è certamente noto il peso molecolare. Questi componento vengono quindi utilizzati per la calibrare l’analisi secondo quanto descritto in precedenza. La calibrazione interna sarebbe potenzialmente più precisa ma, per diverse ragioni tecniche, non è quasi mai applicabile. Comunque, come dettagliato successivamente, la precisione raggiunta con la calibrazione esterna è normalmente più che sufficiente al fine di ottenere analisi estremamente accurate. Tabella 1: Miscela di peptidi standard utilizzata per la calibrazione esterna dello strumento per l’analisi di peptidi in modalità reflectron:
SOSTANZA [M+H]+monoisotopico
Angiotensina II 1046.5420 Angiotensina I 1296.6853 Sostanza P 1347.7361 Bombesina 1619.8230 ACTH frammento (1-17) 2093.0868 ACTH frammento (18-39) 2465.1990 Somatostatina 28 3147.4714

La Figura 5 mostra lo spettro di massa relativo a questa miscela, da cui si può osservare che è presente un picco [M+H]+ per ciascun componente
Fig. 5. Spettro di massa di una miscela di peptidi standard
m/z
Tabella 2: Miscela di proteine standard utilizzata per la calibrazione esterna dello strumento per l’analisi di proteine in modalità linear.
SOSTANZA [M+H]+
Tripsinogeno [M+H]+ 23,982
Proteina A [M+H]+ 44,613

Albumina-Bovina [M+H]+ 66,431
La Figura 6 mostra lo spettro di massa relativo a questa miscela di proteine standard. Si noti che per l’albumina oltre al segnale atteso per la forma [M+H]+ a circa 66.000, si ha un segnale a m/z circa 33.000, corrispondente alla forma [M+2H]2+, che, avendo z = +2, mostra una m apparente che è la metà di quella calcolata per l’albumina
Figura 6: Spettro di massa di una miscela di proteine standard
m/z

Parametri per la valutazione delle prestazioni di uno spettrometro di massa MALDI-TOF
Le prestazioni degli spettrometri di massa TOF sono definite da due parametri: la risoluzione e la precisione di massa. Questi due importanti parametri sono usati per valutare e confrontare le prestazioni di differenti configurazioni strumentali. La risoluzione di massa (m/∆m) è una misura della capacità di uno strumento di produrre segnali separati da ioni di massa simile. È espressa come il rapporto tra la massa m dello ione e la massima ampiezza del segnale ∆m, ricavata misurando la larghezza (in Da) del picco relativo alla proteina a metà altezza del picco stesso. La precisione di massa è una misura dell’errore che si fa assegnando ad una massa un dato segnale. È data dal rapporto tra l’errore dell’assegnazione della massa e la massa reale dello ione ed è frequentemente espressa in percentuale o in parti per milione (ppm). Quindi un errore di 200 ppm significa che se il peso molecolare REALE della proteina è, ad es., 20000, il peso molecolare DETERMINATO SPERIMENTALMENTE dallo strumento sarà compreso tra 19996 e 20004! A puro titolo esemplificativo, al fine di dare una idea quantitativa delle poptenzialità di analisi di questa tecnica, forniamo di seguito i parametri relativi allo strumento Bruker Reflex IV da noi utilizzato attualmente nei nostri laboratori:
• Modalità “linear” :
Risoluzione > 5000 (con frammento ACTH 18-39);
Accuratezza : 78 ppm con sostanza P, 95 ppm con citocromo c.
• Modalità “reflectron” :
Risoluzione > 25000 con somatostatina, >1100 con citocromo c;
Accuratezza: 45 ppm con sostanza P, 100 ppm con citocromo c. Di seguito sono presentati a titolo di esempio due spettri di massa ottenuti rispettivamente (1) da una proteina di recente purificazione di cui si è voluto determinare il peso molecolare e (2) dalla analisi di una miscela triptica di una proteina isolata da elettroforesi bidimensionale (seguendo le diverse metodiche descritte nei vari capitoli precedenti) ed identificata per confronto con informazioni presenti in Banche Dati secondo quanto descritto nel capitolo successivo) .
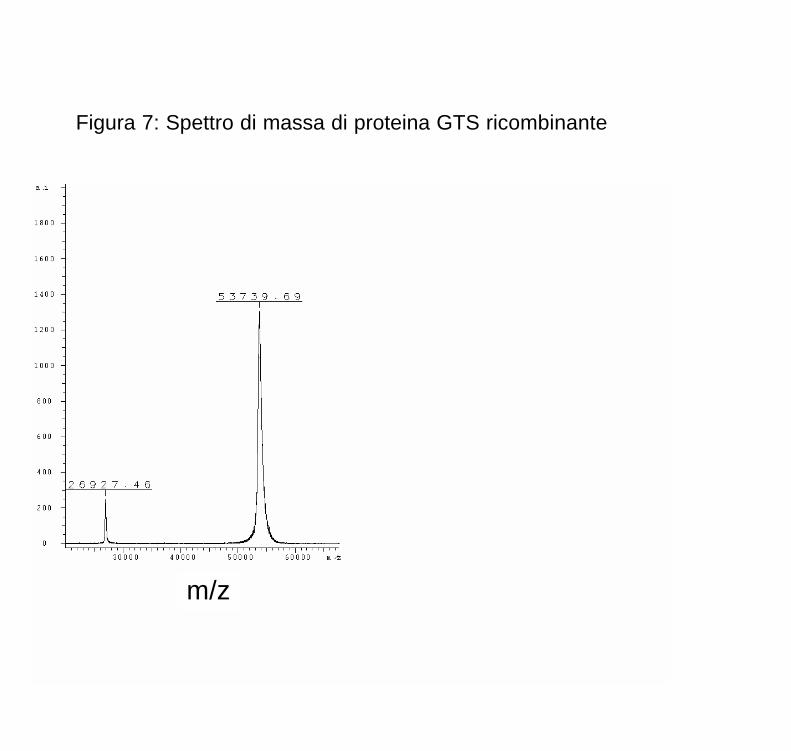
Figura 7: Spettro di massa di proteina GTS ricombinante
m/z
m/z

La Figura 7 mostra lo spettro di massa di una proteina ricombinante, la GTS di M. tuberculosis espressa in E. coli, ottenuta in modalità linear. L’esperimento serviva a verificare se il prodotto ottenuto mediante la tecnologia ricombinante corrispondeva a quanto atteso in base al cDNA codificante la proteina. Si notano nettamente due picchi; il maggiore, corrispondente alla forma [M+H]+ con z = +1, con massa di 53739.69, l’altro corrispondente alla stessa molecola ma con z = +2 (ossia la forma [M+2H]+2). Nel caso specifico qui riportato, la massa attesa dalla sequenza del gene era di 53732, quindi l’accuratezza è di 143 ppm.

Figura 8 Spettro di massa del digerito triptico di una proteina isolata mediante elettroforesi bidimensionale (per l’analisi di questo dato, vedi capitolo successivo)
m/z


















