
3 - T-Space - University of Toronto
109
EVALUATION OF THE HYDROPHOBIC AND OLIGOSACCHARIDE BINDING FUNCTIONS OF THE GM2 ACTIVATOR PROTEIN BY FLUORESCENCE DEQUENCHING ASSAY Natasha Smiljanic-Georgijev Thesis subrnitted in conformity with the requirements for the degree of Master of Science Department of Clinical Biochemistry in University of Toronto @ Copyright by Natasha Smiljanic-Georgijev 1997
Transcript of 3 - T-Space - University of Toronto

EVALUATION OF THE HYDROPHOBIC AND OLIGOSACCHARIDE BINDING
FUNCTIONS OF THE GM2 ACTIVATOR PROTEIN BY FLUORESCENCE
DEQUENCHING ASSAY
Natasha Smiljanic-Georgijev
Thesis subrnitted in conformity with the requirements
for the degree of Master of Science
Department of Clinical Biochemistry
in University of Toronto
@ Copyright by Natasha Smiljanic-Georgijev 1997

National Library 1*1 of Canada Bibliothèque nationale du Canada
Acquisitions and Acquisitions et Bibliographie Services services bibliographiques
395 Wellington Street 395. rue Wellington Ottawa ON K1A ON4 OttawaON K1AON4 Canada Canada
The author has granted a non- L'auteur a accordé une licence non exclusive licence dowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distribute or sell reproduire, prêter, distribuer ou copies of this thesis in microform, vendre des copies de cette thèse sous paper or electronic formats. la forme de microfiche/film, de
reproduction sur papier ou sur format électronique.
The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts £kom it Ni la thèse ni des extraits substantiels may be printed or othewise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation.

TABLE OF CONTENTS
........................................................................................ AB STRACT v ... .......................................................................... ACKNOWLEDGMENT vri ... ................................................................................ AB B REVIATIONS viu
.................................................................................. PUB LICATIONS ix
CHAPTER 1 GENERAL INTRODUCTION
........................................................................ 1.1. GM2 Gangliosidosis 2 ............................................ 1.2. Structure of the HEXA and HEXB Genes. - 2
1.3. Hexosarninidase Isozymes ................................................................ - 5 1.4. The Interaction Between the Activator . GM2 Ganglioside and ........................ 6
............................................................................... Hexosaminidase A - 6
1 .5 . Genomic Structure of the GM2A Gene Encoding the GM2 Activator Protein ...... - 9
1.7. AB Variant Form of GM2 Gangliosidosis ................................................ 14
1.8. Other Lysosomal Sphingolipid Activator Proteins ........... .... ................... 17
1.9. Other Possible Functions of the GM2 Activator Protein ................................ 18
................................................................................ 1.10. Gangliosides 19
1.10.1. Ganglioside Biosynthesis ............................................. 22
............................................... 1.10.2. Ganglioside Catabolism 25
.......................................................................................... References 26
CHAPTER n DEVELOPMENT OF THE FLUORESCENCE DEQUENCHING ASSAY FOR STRUCTURE / FUNCTION STUDIES OF THE G M ~ ACTIVATOR PROTEIN
2.1 Introduction ................................................................................... 34
2.2 Materials and Methods ...................................................................... - 38 ................................................................. 2.2.1. Materials 3 8
2.2.2. Production of the His6-GM2 Activator Fusion Protein from E-Coli
.................................................................................. 3 8
2.2.3. Preparation of Phosphatidylcholine Containing Large Unilamellar
.......................................................................... Vesicles -42
2.2.4. Preparation of R- 18 labeled liposomes .............................. -42

.................................... 2.2.5. Fluorescence Dequenching assay -42
....................................................................................... 2.3. Results -43
2.3.1. Fluorescence dequenching of R- 18 labeled liposomes in the presence of
recombinant human GM2 activator protein ............. .. ................ 43
2.3.2. Detemination of the optimal time course range for the fluorescence ............................................................. dequenching assay -50
2.3.3. Detemination of the optimal GM2 activator protein amount for the
fluorescence dequenching assay ................ .. .................... 50
2.3.4. Determination of the optimal pH ..................................... -59
................................................................................... 2.4. Discussion -59 .......................................................................................... References 61
CHAPTER nI EVALUATION OF THE HYDROPHOBIC AND OLIGOSACCHARIDE BINDING FUNCTIONS AND IDENTIFICATION OF A SECRETORY FORM OF THE G M ~ ACTNATOR PROTEIN
................................................................................. 3.1. Introduction -64 ..................................................................... 3.2. Materials and Methods -67
3.2.1. Materials .......................... .,.. ................................... 67
3.2.2.Expression and Purification of His6-GM2 Activator Protein Constmcts
from E . Coli ...................................................................... 68
3.2.3.The Expression and Purification of the His6-Activator Containine a ....................................................... Cys 138Arg Substitution -68
..................................... 3.2 4 . Fluorescence Dequenching Assay 69
3.2.5 Analysis of the Distribution of Activator Molecules with a High Mannose
................................. or a Complex Type Oligosaccharide Moiety 69
3.2.5.1. Isolation of a Semi-Purified Sarnple of Newly Synthesized Activator from
Normal Human Fibroblasts ............................................................. 69
........................................................ 3.2.5.2. Glycosidase Digestion 70
........................................................ 3.2.5.3. Western Blot Analysis 71
3.3. Results ........................................................................................ 71
3.3.1. Determination of the Recombinant Hurnan GM2 Activator Protein .............. Binding Affinity for Various Glycolipids and Gangliosides -71
3.3.2. Analysis of the Hydrophobic Function of Different Recombinant
Human GM2 Activator Protein Constmcts .............. .... ............ 81
3.3.3. Hydrophobic Function of the Activator Protein Containhg a Point Mutation

Linked to the AB Variant Form of G M ~ Gangliosidosis by Ruorescence Dequenching
.............................................................................. Assay 86
3.4. Discussion.. ................................................................................. -86 References ......................................................................................... -92
CHAPTER N FUTURE WORK
4.1. X-ray Crystallographic Studies of the GM2 Activator Rotein ......................... 96 4.2. Determination of the R-18 / Ganglioside Binding Site of the GM2 Activator Protein by
Mu tagenesis ....................................................................................... -96
4.3. Identification of the Hexosaminidase A Binding Site of the GM2 Activator Protein Through the Expression of the Mouse / Human Fusion Protein ........................... -97
R e f e r e n c e s .......................................................................................... 98

ABSTRACT
G M ~ activat~r protein is a substrate specific cofactor for degradation of GMZ ganglioside by
phexosaminidase A. It solubilizes individual molecules of the ganglioside by interacting with both
its hydrophilic oligosaccharide and hydrophobic ceramide moieties. However, the specificity of
binding is pnmarily determined by the oligosaccharide moiety of the glycolipid. Thus, the activator
protein contains at least three functional elements; a hydrophobic binding pocket, an oligosaccharide
binding site, and an area that interacts with hexosarninidase A. Mutations in the gene encoding the
activator result in the AB-variant form of G M ~ gangliosidosis.
The goal of this thesis is to evaluate the hydrophobic and oligosaccharide binding functions of
the G M ~ activator protein. Recentiy, the investigation of endosomeAysosome fusion hy fluorescence
dequenching assay, suggested that G M ~ activator protein c m act as a transfer protein of the
fluorescent lipid probe. octadecylrhodamine, between egg phosphatidylcholine (PC) liposomes. as
weil as isolated endosornes and lysosomes. Based on these findings rny first objective was to develop
a fluorescence dequenching assay that could be used to evaluate the hydrophobic binding function of
the activator protehi. This assay was based on the concentration dependent fluorescence dequenching
of the fluorescent Lipid probe in presence of the G M ~ activator protein. The optimal time course was
determined to be from 5th to 10th minute after the initiation of the assay (first five minutes were
required for fluorescence stabilization). The optimal amount of the activator protein and pH used in
this assay, were found to be from 0.75 pg to 4 pg and pH 5. respectively.
The second objective of this thesis was to determine the activator protein binding affinity for
various glycolipids and gangliosides by the fluorescence dequenching method. 1 determined the
degree to which various glycolipids could inhibit the transport of the octadecylrhodamine. between
liposomes. by the activator protein. The levei of inhibition produced by each glycolipid was used to
charactenze the oligosaccharide-binding specificity of the activator: G M ~ » G ~ l b » G ~ l 2 -2
G M ~ > GA^-

The fluorescence dequenching assay was also used to evaluate the hydrophobic function of the
C-terminal part of the G M ~ activator protein. 1 investigated the functionality of three truncated
activator protein consmcts, lacking different number of C-terminal amino acids (36, 11 and 9 amino
acids, respectively). AU three constructs were inactive in releasing fluorescence, compared to the wild
type activator, indicating that they lacked a functional hydrophobic binding site. Thus, it is possible
that the C-terminal part of the activator protein could be involved in the ganglioside binding function.
Finally. the hydrophobic hnction of the activator protein containing a point mutation Linked to
the AB variant f o m of G M ~ gangliosidoses (T412c (Cp 138Arg )) was analyzed by the fluorescence
dequenching assay. This protein caused an increase of fluorescence which was 96% of that caused
by the wild type. This function was suongIy inhibited by Gh.12 ganglioside, suggesting that the
mutated activator had a fully functional ganglioside binding site. These data suggest that ~~s~~~ may
not be involved in the ganglioside binding function of the activator protein. but only in the interaction
with Hex A, possibly through the formation of disulfide bond with another cysteine.

ACKNOWLEDGMENT
The work presented in this thesis was done in the Division of Neurosciences at the Hospital
for Sick Children, Toronto, under the supervision of Dr. Don Mahuran. 1 would like to extend rny
sincere gratitude to Dr. Mahuran for allowing me the opporninity to improve my scientific knowledge
under his continued guidance and support. 1 also wish to acknowledge Dr. Joan Boggs and Dr. John
Callahan for their advice and help over past two years, as well as Dr. Fred Keeley for his participation
in evaluating my work.
1 am very indebted to people in the laboratory for their friendship and assistance. 1 would like
to thank Mr. Roderic Tse for helpful discussions and friendly collaboration. 1 very much appreciate
advises and assistance of Mrs. Euijung JO. Mr. Jude Pereira a ~ d Dr. Brigitte Rigat. Special thanks to
Ms. Amy Leung and Mrs. Godha Rangaraj for their great technical assistance. 1 would also like to
acknowledge Dr. Sunqu Zhang, Dr. Benny Chen, Mrs. Irene Warren. Mrs. Marie-Anne
Skomorowski. Mrs. Kathleen Sparacino. Mrs. Caroline Bruce and al1 members of Dr. Cecil Pace-
Asciak's and Dr. FIavio Coceani's laboratories for making the Neurosciences an enjoyable place to
work.
Most of al1 1 rhank my husband, Zvonimir Georgijev, for his love and support during the
time. as well as our two wonderful sons. Nikola and Peter. for making my Iife joyous and
worthwhile. 1 specidly acknowledge families Smiljanic and Georgijev whose help and continuous
encouragemznt made the beginning of my Life and work in Canada more successful and easier.

LIST OF ABBREVIATIONS
Act 183 Act 185 cDNA Cer CHO CMF' EDTA ER FCS Ga1 GaiNAc Glc GlcNAc GM2A GM2A P GSL HEXA W E X B Hex rPTG kDa Man MEM mRNA M-6-P MPR 4-MUGS NeuNAc Ni-NTA a3 PC-LUV PCR R-18 S A P SDS-PAGE SFM UDP
muicated activator protein ma& by point mutation at codon 183 mmcated activator protein made by point mutatior; at codon 185 complementary DNA ceramide Chinese hamster ovary cytidine-5'-diphosphate ethylenediaminetetraacetate Endoplasmic reticulum fe tai calf senun Gala tose N-acetylgalac tosamine Glucose N-acetylglucosamine gene encoding G M ~ activator protein pseudogene related to GMU g~ycosphingolipids gene encoding a-subunit of Hex A
gene encoding ksubunit of Hex A
isopropyl- l -thio-PD-galactoside kdodaltons mannose minimum essential media messenger RNA mannose-6-phosphate mannose-&phosphate receptor Qmethylumbelliferyl P-N-acetylglucosarnine 6-sulfate N-ace ty lneuraminic acid (sialic acid) nickel-nitrilo tri acetic acid optic density phosphatidilcolin conraining large unilarnellar vesicle polymerase chah reaction octadecilrhodarnine B chloride sphingolipid activator protein sodium dodecil sulphate polyacrylamide gel electrophoresis semm free medium uridine-5'-monophpsphate

PUBLICATIONS
Natasha Smiijanic-Georgijev. Brigitte Rigat, Bei Xie, Wei Wang and Don J.
Mahuran. "Characterization of the Affinity of the G m Activator Protein for Glycolipids by
Fluorescence Dequenching Assay" Biochim.Biophys.Acta, in press.

C H A P T E R 1
GENERAL INTRODUCTION

The G M ~ gangliosidoses are a group of inherïted disorders caused by excessive
intralysosomal accumulation of ganglioside G M ~ and related glycolipids. particularly in neuronal
cells. These disorders result from defective G M ~ catabolism, which physiologicaIIy involve the
action of a lysosomal enzyme, f3-hexosaminidase A (Hex A). which is composed of two subunits, an
a and a p, and its substrate specific CO-factor the G M ~ activator protein. Since the a and the B subunit of Hex A are encoded by two unlinked structural genes. HEXA and HEXB, G M ~
degradation requires the participation of three geneticaily distinct polypeptides (Fig 1.1 .) (reviewed in
(Fürst and Sandhoff 1992)). Mutations in any of the three genes encoding these proteins may give
rise to G M ~ gangliosidosis. The three forms of GMZ gangliosidosis are; Tay-Sachs disease (a
defects). Sandhoff disease (P defects), and AB variant f o m of G M ~ gangliosidosis ( G M ~ activator
protein defects) (reviewed in (Gravel et al. 1995). Tay-Sachs disease occurs most frequently in
Ashkenazi Jews (1/3600 birth) (Kaback et al. 1977). primarily as a result of two very frequent
mutations (Gravel et al. 1995). To date. nearly 60 H E M gene mutations leading to this disease in
various populations have been identified. Sandhoff disease and the AB variant form of gangliosidosis
are rarer than Tay Sachs disease. with a total of 17 HEXB mutations and two GMZA mutations
k i n g Linked to disease states (reviewed in (Gravel et al. 1995)).
1.2. STRUCTURE OF THE HEXA A N D HEXB GENES
The HEXA gene maps to chromosome 15q23-q24 (Nakai et al. 1991) while the HEXB gene
maps to chromosome 5q13 (Bilcker et al. 1988). The HEXA gene is 35 kb long and contains 14
exons (Proia and Soravia 1987). The HEXB gene has about 45 kb and also contains 14 exons
(Neote e t al. 1988; Proia 1988). The structures of HEXA and HEXB genes show a striking degree
of homology in both the number and the placement of exon / intron junctions. As well. a cornparison
of the deduced primary sequences from their cDNAs, reveals an overall57% identity.

Figure 1.1 .:
The ~hexosaminidase gene system (from (Grave1 et al. 1995)).

Gene
Polypeptide
aa Hex S
Su bstrates
HEXA HEXB Chr 1 Sq23-24 Chr Sq13
4 Hex A
QQ Hex 8
GMZA Chr 5q32-33
Physiotog ic Glycosaminogl ycans? Glycoproteins G!ycoproteins Oligosaccharides OIigosaccharides GIycosaminoglycans GIycosarninoglycans GlycoIipids Gl ycolipids
I ~ a n ~ l i o s i d e GM~\ - cornplex formation

These data suggested, that the HEXA and HEXB arose from a common ancestor (Proia 1988;
Komeluk et al. 1986)- Thus, functional domains are Lkely to be conserved within aligned structures
of the two subunits. This hypothesis has b e n confirmed expetimentally (Brown et al. 1989: Brown
and Mahuran 1993; De Gasperi et ai. 1996).
1.3. HEXOSAMINIDASE ISOZYMES
PN-acetylhexosaminidase. Hex, is a lysosornal hydrolase that cleaves texminal non-reducing
P- 14-Iinked N-acetylglucosamine (GlcNAc) and N-acetylgalactosamine (GalNAc) residues frorn
oligosaccharides. glycolipids, gangliosides* glycoproteins and glycosarninoglycans. There are three
human P-hexosaminidase isozymes which are made up of al1 possible dimeric combinations of a
andor p subunits: A (ap) , B (PP) and S (aa) (Srivastava and Beutler 1973; ikonne et al. 1975).
Only dimer forms of Hex are functional, however each subunit is believed to contain one potentially
active site or two partial active sites. Active sites associated with eilher a or B subunits are able to
hydrolyse some sarne neutral artificial (Hou et al. 1996) and natural substrates (Mahuran et al. 1985).
But only the catalytic site associated with the a subunit c m hydrolyse negatively charged substrates,
e - g . CMUGS (Bayleran e t al. 1984). P-linked glucosamine 6-sulphate containing
glycosaminoglycans (Kresse et al. 1981). and the most important G M ~ ganglioside (Meier et al.
1991). Recently. based on the structural homology between a and P subunits and the common
evolutionary origin of the H E U and HEXB genes. the functional areas within two subunits were
located by cellular expression of a@ fusion proteins joined at adjacently aligned residues (Tse et al.
1996). It was concluded that: a) The least homologous amino terminal sections of a and D
polypeptides are only needed for proper folding in endoplasmic reticulum (ER). b) The most
homologous middle sections contain the substrate binding / active sites. c) The carboxy terminal
sections are likely involved in subunit-subunit interactions.

1.4. TEE INTERACTION B E T W E E N THE ACTIVATOR, G M ~ GANGLIOSIDE AND
Hex A can not directly attack the membrane associated substrate (Conzelmann and Sandhoff
1978) but can degrade G M ~ in the activator-GM~ complex in vivo (Conzelmann et al. 1982), or in
vitro in the presence of detergent. This suggests that the activator acts primarily as a substrate
specific CO-factor of Hex A. instead of " activating" the enzyme (Sandhoff et al. 1989). The G M ~
activator protein itself, appears unable to penetrate the liposomal membrane, as only lipid moIecules
on the outer leaflet of the membrane are accessible to the activator (Conzelmann et al. 1982). It binds
G M ~ and "presents" it to Hex A for degradation (Sandhoff et al. 1989). Figure 1.2. presents
possible models of the activator assisted G M ~ hydrolysis by Hex A (reviewed in (Fürst and Sandhoff
1992)).
The activator interacts with both the hydrophilic oligosaccharide and hydrophobic ceramide
moieties of gangliosides. The hydrophobic binding site for the ceramide portion of gangliosides has
ken suggested to be composed of a pocket forrned by amphiphilic a helices predicted from the arnino
acid sequence of the activator (Fürst et al. 1990). The spectmm of glycolipids that interacts with the
activator is primarily determined by their oligosaccharide moieties. In vitro binding studies have
indicated that the terminal N-acetylgalactosamine (GalNAc) and interna1 sialic acid (NeuAc) residues
of GMZ play an important role in deterinking binding specificity, Le. GM2>> GM 1> GD la= GM3=
GA2. The resulting 1 to 1 activator: ganglioside complex must then specifically interact with Hex A
(reviewed in (Sandhoff et al. 1995))-
Studies conceming the interactions of the G M ~ activator protein with Hex A suggest that the
molar ratio of Hex A to the activator 1 ganglioside complex neccssary for obtaining the maximal
hydrolysis of G M ~ is close to 1: 1 (Li et al. 198 1). The degradation of CMUGS (a negatively
charged artificial substrate that is bound by the active site associated with the a subunit) by Hex A
(ap) and Hex S (aa) cm be competitively inhibited by the G M ~ activator protein, indicating that the
a subunit of Hex A also contain an activator-binding site (Kytzia and Sandhoff 1985). As the

Figure 1.2.:
Two models presenting the interaction of G M ~ activator with Hex A (from (Fürst and
Sandhoff 1992)).
Model 1: The formation of G M ~ , G M ~ activator and Hex A complex can take place on the
lysosomal membrane radier than in the free solution, and the function of the G M ~ activator is to lift
G M ~ a few angstroms out of the membrane which eliminates the stenc hindrance of adjacent lipid
molecules and gives the enzyme access to the Blinked GaNAc residue.
Model 2: The G M ~ activator vansports G M ~ from membrane to free solution of the lysosome,
presenhg it to Hex A for hydrolysis.
Either or both models may exist-


activator does not promote degradation of G M ~ by Hex S. it is possible that Hex S does not bind to
the GMZ activator 1 G M ~ complex in the correct orientation and that the subunit of Hex A may also
interact with the activator (Kytzia and Sandhoff 1985) .
1.5. GENOMIC STRUCTURE OF THE G M 2 A GENE ENCODING THE G M ~ ACTIVATOR
PROTEIN
Recently, the fful length cDNA clones encoding the GMZ activator protein have been isolated
by ourselves and others (Xie et al. 199 1 ; Klima et al. 1991 ; Nagmjan et al. 1992). The GM2A gene
has been rnapped to chromosome 5q32-33, while a pseudogene. CMZAP, has been mapped to
chromosome 3 (Xie et al. 1992; Swallow et al. 1993). The structure of -95% of the human GMZA
gene has been characterized and four exons idenûfied. The 2.4 kb cDNA codes for a prepro-protein
of 193 arnino acids. The signal peptide, required for the entry into the rough endoplasmic reticulum
(23 amino acids) and the mature protein (162 amino acids) are connected by 8 amino acids which are
removed during maturation in the lysosome (Fürst et al. 1990). The deduced primriry sequence. also
contains one consensus site for Asn-linked glycosylation (Asn63-Val-Thr).
A second alternatively spliced mRNA product containing exons 1-3 and intron 3 has been
identified (Fig. 1.3.) (Nagarajan et al. 1992). Due to the presence of a STOP codon early in the
retained inuon 3 sequence, the product of the alternatively spliced mRNA is a truncated form of the
activator missing residues 142-193 (counting from the initial Met residue) and containing an additional
three residues encoded by the inuon 3, Val-Ser-Thr.

Figure 1.3.:
Gene structure of G M ~ activator protein and G M ~ A protein (from (Wu et al. 1996)).
El, E2, E3 and E4 represent exon 1, exon 2, exon 3 and exon 4, respectively. I l , 12 and 13
represent intron 1. intron 2 and intron 3. respectively. The word "stop" means stop codon. VST is
the last three amino acids encoded by intron 3. The sizes of the gene fragments are not in the exact
proportion.

G , , activaror inRNA %,A
1 El 1 ES i E3 1 E4 1 1 El ES 1 E3 1 13 1
G,, ac~ivaior protein
[ ~ l r E2 1 E3 1 E4 1 CM,, proiein

The G M ~ activator protein as well as the a and i3 chains of hexosarninidase A are
glycoproteins synthesized on polysomes attached to the rough endoplasmic reticulum (ER). To enter
the rough ER glycoproteins require a signal peptide (residues 1-23 in the G M ~ activator protein). The
signal peptide is cleaved by signal peptidase in the lumen of the ER. This event may be followed by
the addition of an oligosaccharide c h a h to the asparagine residues contained in the consensus
sequence (Am-X-Ser/Thr). This glycosylation step involves the en block transfer of large preformed
oligosaccharide. GalNAc2 Mang Gk3, from a lipid-linked intennediate to the nascent polypeptide
(Komfeld and Kornfeld 1985). Before exiting the ER glycoproteins lose their Glcg groups from the
oligosaccharides and fold to their near native conformation. In some cases, like Hex. oligomerization
must also occur; however, the activator protein is a monomer. A group of resident ER proteins,
chaperones, bind to the elongating nascent polypeptide, preventuig aggregation of the polypeptide and
accelerate its correct folding. Another chaperone. disulfide isomerase, assists in the correct placement
of disulfide bonds (Hendrick and Hartl 1993). If nascent polypeptides are not released from
chaperones, e-g. due to mutations, their degndation rate is increased.
After the newly synthesized glycoproteins are properly folded, they enter Ihe cis Golgi where
transport continues unidirectionally via bulk flow. For most lysosomal proteins. mannose-6-
phosphate (M6P) markers are added to one or more high mannose type oligosaccharides. The M6P
recognition marker is generated by the sequential action of two Golgi enzymes. First, N -
acetylglucosamine-phosphotransferase transfers N- acetyIglucosamine-1-phosphate from the
nucleotide sugar uridine diphosphate-N-acetylglucosamine to selected mannose residues on lysosomd
enzymes. to $ive rise to a phosphodiester interrnediate. nien. N- Acetylglucosamine phosphodiester
glycosidase removes N-acetylglucosamine residue to expose the recognition signal (Lang et al. 1984).
The phosphotranspherase is the enzyme defective in patients with 1-ce11 disease. This disease is
characterized by the tissue specific intracellular deficiency of many lysosomal proteins which are
elevated in the plasma of the patients. Fibroblasts from these patients fail to phosphorylate mannose

residues on their newly synthesized lysosomal proteins and secrete a large percentage of thern into the
culture medium, as a precursors. (Hasilik and von Figura 198 1; Burg et al. 1985; Varki et al. 198 1).
The phosphorylation of lysosomal glycoproteins specifically targets them to the lysosome
through the interaction with one of two mannose-6-phosphate receptoa (MPRs) in the trans Golgi
network (Griffith et al. 1988). MPRs are tram-membrane proteins which c m be concentrated in
clathrin coated vesicles on the membrane of the tram Golgi network or the plasma membrane by
adaptor proteins (Glickman et al. 1989). Adaptors recognize specific protein motifs in the
cytoplasmic domain of transmembrane proteins and promote polyrnerization on clathrin triskelions
ont0 the membranes (Pearse and Robinson 1990). The receptor-ligand binding in the Golgi diverts
lysosomal proteins away from the default route followed by secretion proteins containing complex
oligosaccharide structures. After these interactions. the membrane of the clathrin coated vesicles buds
from the trans Golgi membrane forming srnall cytosolic vesicles. These vesicles translocate
lysosomal glycoproteins into the late endosome where acidic pH causes uncoupling of the MPRs and
ligands. Ligands are then transported via vesicles to lysosomes or the late endosome fuses with or
becomes a mature lysosome (Croze et al. 1989). The recepton recycle back to the Golgi.
In the lysosome or late endosome a series of proteolytic and glycosidic processing events
occur to the pro-Hex isozymes forming their mature subunit stnicturcs (Mahuran et al. 1988; Stirling
et al. 1988; Hubbes et al. 1989; O'Dowd et al. 1988; Mahuran 1990). Proteolytic processing of the
single 67 kDa pmpolypeptide chain results in the generation of the pp ( 1 1- 14 kDa). Pb (22-24 kDa)
and Pa (26-28 kDa) chains. Similarly, the 65 kDa a propolypeptide results in genention of the a p (7
kDa) and a m (56 kDa) chains. comprising the mature P and a chains. respectivcly. Each set of
mature a or are held together by disulfide bonds in the mature subunits.
Transportation of the G M ~ activator protein to the lysosome is still not completely understood.
Two reports have suggested that the activator is transported to the lysosome via MPR. First, there
was a 2.5 fold increase in the amount of activator in the s e m of a single 1-ce11 patient (Banerjee et al.
1984) and secondly. there was a similar increase in the secretion of the activator from normal
fibroblasts grown in the presence of NH4CI (which prevents acidification of both endosornes and

lysosomes. causing the newly synthesized precursors to be treated as secretory proteins) (Burg et al.
1985). However. these data are far from conclusive a s the secretion of other MPR-targeted enzymes
are increased 10-50 fold with a concomitant loss of intrace11ular enzyme in b o t . circumstances (Creek
et al. 1983; Wiesmann et al. 197 1))-
After entering the lysosome, the pro G M ~ activator protein. 24 D a . is processed by
proteolytic and glycosidic enzymes, foming the 22 kDa mature form. The pro activator can be
precipitated from the culture medium, while only mature form is detected in cells. suggesting a rapid
processing compared to the low biosynthetic rate (Burg et al. 1985). The molecular mass of the
polypeptide c h a h without its single oligosaccharïde is 17.6 kDa (Fürst et al. 1990). The mature G M ~
activator has eight cysteine residues in its 162 amino acid polypeptide chain. Presumably the
formation of 4 cystines. from these cysteines. is responsible for the hert stability of the protein (Fürst
et al. 1990).
AB variant form of gangliosidosis is extremely rare and is caused by mutations in the gene
encoding G M ~ activator protein. To date, four mutations have k e n reported by ourselves and others;
a T4 12 to C (Cys 138 to Arg ) (Xie et al. 1992), a G506 to C (Arg 169 to Pro) (Schroder et al. 1993),
a deletion ~ ~ ~ 2 6 * - 2 6 4 (deletion of ~ ~ ~ 8 8 ) . a deletion ~ 4 1 0 (frarne shift) (Schepers et al. 1996).
These mutations were associated with the infantile acute form of the disease. This form of G M ~
gangliosidosis. AB variant. is clinically indistinguishable frorn the infantile onset of Tay-Sachs and
Sandhoff diseases. The symptoms, involving the l o s of motor skills, ophthalmologic problems and
the increase of seizure activity, begin in the first 3 to 5 months of life and progress rapidly with a fatal
ending between 2 to 4 years of age. AU G M ~ gangliosidoses are histopathologically charactenzed by
the accumulation of G M ~ ganglioside, mostly affecting neuronal lysosomes. which appear by electron

Figure 1.4.:
Concenvic lamellar bodies in G m gangliosidosis (from (Agamanolis 1995)).


microscopy as numerous unilarnellar bodies, consisting of dense concenttic membranes (Fig. 1.4.)
(reviewed in (Agamanolis 1995)). However, in the AB variant f o m of G M ~ gangliosidosis, changes
including prominent inclusions in glial ceiis and large conglornerates of lipid inclusions, are found to
be significantly different from those in Tay-Sachs and Sandhoff diseases. These differences indicate
that the activator may have some other, as yet uncharacterised. function(s) Le. the involvement in the
general glycolipid transportation.
G M ~ activator is one of five known lysosomal sphingolipid activator proteins (SAPs)
(reviewed in (Fürst and Sandhoff 1992)). Four of these, sap A-D. are encoded by a single gene
mapped on chromosome 10. They enter the lysosome as a single 65-73 kDa precursor chain. The
large precursor, prosaposin, is processed into four - 13 kDa polypeptide chains that share a fair
degree of structurai hornology. Sulphatase activator. Saposin-B or SAP- 1, activates the hydrolysis of
cerebroside sulphate, GM 1 and globotnaosylceramide by arylsulphatase A, B-galactosidase and a-
galactosidase, respectively. Glucosylcerarnidase activator, Saposin-C or SAP-2, stimulates the
hydrolysis of glucosylceramide, galactosylcerarnide and sphingornyelin by O-glucosylceramidase. O-
galactosidase and sphingomyelinase, respectively. Other two potential activator proteins are Saposin-
A and Saposin-D. In vitro. Saposin-A activates glucosylcerarnidase and galactosylceramidase, while
Saposin-D shows some stimulatory effect on the degndation of cerarnide in vitro and in vivo.
h comparison to other SAPs. the G M ~ activator protein is unique in several aspects: a) it is
encoded by an unrelated gene on chromosome 5 (Xie et al. 1992), b) it shares no significant deduced
primary structure homology with Sap A-D (Schroder et ai. 1989). c) it functions as a monomer (sap
A-D are hornodirners) and d) its only proven in vivo function is as a substrate specific cofactor for the
degradation of G M ~ ganglioside by Hex A, a rolc that can not be filled by any of the other SAPs
(Sandhoff et al. 1995).

Recently, in addition to its role as a lysosomal precursor of sap A-D. prosaposin was found to
exist as a secretory protein in human milk, cerebrospinal fluid and seminal plasma (Hiraiwa et al.
1993). It was further demonstrated that prosaposin can bind gangliosides with high affinity and
facilitate their transfer from micelles to membranes (Hiraiwa et al. 1992). Taken together, these in
vitro and in vivo studies of prosaposin, suggest it as a possible intraceilular ganglioside binding and
nansfer protein. Furthemore. prosaposin was identified as a neurotrophic factor, stimulahg neurite
outgrowth by binding to a high-affinty receptor (O'Brien et al. 1994).
1.9. OTRER POSSIBLE FUNCTIONS OF THE G M ~ ACWATOR PROTEIN
The only proven in vivo function of the G M ~ activator protein is to stimulate the breakdown
of G M ~ to G M ~ ganglioside by Hex A (Conzelmann et al. 1979). In vitro. it also promotes the
conversion of G M ~ to GAZ by clostridial sialidase, the hydrolysis of GA^ by Hex A and the release
of Ga1 from GM 1 by B-galactosidase when added in high concentrations (Kytzia and Sandhoff 1985;
Wu et al. 1994). As weI1, in the absence of Hex A, the activator c m act in vitro as a general
sphingolipid transport protein. The rate of the ganglioside transfer between liposomes is: G M b
GM2> GDla>> GM3= GA2 (Conzelmann et al. 1982). Thus, one of the most abundant brain
gangliosides. GM 1, is transferred at a preferential rate. Furthemore, the activator was also shown to
transport a fluorescent lipid probe between labeled and unlabeled liposomes (Kuwana et al. 1995).
Thus, as with prosaposin (Hiraiwa et al. 1992). it is possible that the activator can act in vivo as a
generd glycolipid sorting / transport protein. The retention of the activator's transport function at
neutral pH in vitro (Conzelmann et al. 1982; Kuwana et al. 1995) also supports this hypothesis.
Recently, the function of the G M ~ A protein produced by alternative splicing of the activator
mRNA. which is truncated at residue 142 and contains 3 unrelated amino acids, was investigated (Wu
et al. 1996). This protein, synthesized in bactena and refolded. was reported to retain its hydrophobic
binding pocket and NeuAc recognition site based on; a) its ability to stimulate the hydrolysis of
to GA^ by clostridial sialidase, and b) its inability to stimulate the removal of NeuAc from the

oligosaccharide of the G M ~ ganglioside alone. i. e. without the ceramide moiety , in the presence of the
same enzyme. The GM~A protein also lacked the ability to stimulate the removal of the GaiNAc
residue from by Hex A. suggesting it lacked either the GalNAc recognition site or its domain for
interacting with Hex A.
Glycosphingolipids (GSL) are amphiphilic molecules that constitute the basic Iipid core
structure of ce11 membranes. Whereas they are present at relatively high concentrations in neuronal
plasma membranes, they are also present in the plasma membrane of essentially ail other mammalian
cells. The essential molecular features of glycosphingolipids are a hydrophobic backbone, ceramide
(Cer). which consists of a long chah aliphatic amino alcohol (sphingosine) which is attached to a fatty
acid via an amide linkage and a polar carbohydrate head group which protmdes into the extracellular
environment. Neutra1 glycosphingolipids have one or more neutral sugars, covalently attached via a
glycoside bond to the ceramide. Gangliosides are a group of acidic glycosphingolipids, containing in
addition to the neutral sugar. one or more N-acetylneurarninic acids (NeuAc or sialic acid) (reviewed
in (Hoekstra and Kok 1992; Ledeen and Yu 1982)). The major gangliosides in the body are GM 1,
GD l a and GD 1 b. Others, G M ~ . G M ~ and GT, are intermediates in both the synthesis and
degradation of these more complex gangliosides and consequently are found in lower concentrations.
G M ~ ganglioside (Fig 1.5.) consists of a cerarnide. made of sphingosine and stelinc acid, and the
teuasaccharide made of glucose (Glc), galactose (Gal), N-acetylneuraminic acid (NeuNAc) and N-
acetylgalactosarnine (GaiNAc) (Zeller and Marchase 1992).
The precise biological role(s) for gangliosides has yet to be elucidated. However. recent data
has suggested some important roles that gangliosides may play in the physiologie operations of the
nervous system, in particular that of brain. Their potential roles are in the developmental. ce11
adhesion and signal transduction processes (reviewed in (Zeller and Marchase 1992)). Sorne

Figure 1.5.:
Structure of G M ~ ganglioside.

GalNAc G al GIc ceramide

gangliosides have k e n identified as binding sites on ce11 surfaces For viruses and bacterial toxins.
For example, the cholera toxin subunit B binds to cells specifically through GM 1. allowing the A 1
subunit of the toxin to enter the cells and activate adenylate cyclase (Fishman et al. 1993).
Furthermore, the levels of individual gangliosides are significantly changed in some human tumors.
e.g. the disialoganglioside GD^ is synthesized in abundance by pnmary untreated neuroblastoma and
can be detected in the plasma of patients with this type of tumor (Valentino et al. 1990; Sariola et al.
1991). Finaily, the growth of ectopic dendrites of cortical pyramidal neurons has heen shown to
specifically correlate wiih the arnounts of accumulated G M ~ , suggesting an unique in vivo function
for G M ~ ganglioside (Siegel and Walkley 1994).
Ganglioside biosynthesis starts in the ER where the ceramide portion is synthesized frorn
serine and paimitoyl CoA. After addition of the amide-linked fatty acid. the c e m i d e is transferred to
the Golgi apparatus by an as yet unidentified mechanism. Ceramide is glycosylated in a stepwise
manner by the transfer of the individual sugar from the respective uridine-5'-diphosphate (UDP)
derivatives and sialyl residues from the cytidine-5'-monophosphate (CMP) - Neu NAc. First step of
glycosphingolipids synthesis is the linking of glucose to cerarnide and formation of glucosyiceramide
(GlcCer) in the presence of glucosyltransferase. The coupling of galactose to GlcCer yields
lactosylceramide (LacCer). the precursor of more complex glycosphingolipids. This reaction is
catalyzed by galactosyltransferase. The sequential addition of s id ic acid residues to the growing
oligosaccharide chain. yielding G M ~ , GD^ and more complex ganglioside is catalyzed by
sialyltransferases which exhibit relatively high levei of substrate discrimination. On the other hand,
there is a single N-acetylgalactosarninyltransferase that catalyses the synthesis of GAZ. G M ~ . GD2
and G T ~ by the addition of the third neutral sugar GalNAc. Similarly, the synthesis of terminal
carbohydrate structures s h m d by other glycolipids is aiso catalyzed by this set of enzymes.

Figure 1-6.:
Degradation pathway of ganglioside G M ~ in lysosome showing the type of ganglioside. its
structure and the glycohydrolases involved in its breakdown.

Gai-GalNAc-Gai-GIc-Ceramide
4 I
G M r ganglioside NANA
Gal GMi ganglioside O-galactosidase
GaINAc-G 1-Glc-Ceramide a GM2 ganglioside
Gaf NAc
GM3 ganglioside
NANA
lactosylceramide
Gal
glucosylceram ide
Glc
1 NANA
1 f3-hexosaminidase A
Gal-Glc-Ceramide i
NANA
neuraminidase
Gal-Glc-Ceramide i
"1 "B-galactosidase"
glucocerebroside O-glucosidase
Ceramide
i ceramidase
fatty acid + sphingenine

After their synthesis in the Golgi, gangliosides are transported to the plasma membrane by vesicular
flow and anchored to the outer leaflet of the membrane by their ceramide moieties, with their
oligosaccharides extending into the extracellular space (Van Echten and Sandhoff 1993). Despite an
abundance of studies, detailed locdization of ganglioside biosynthesis as well as mechanisms of
intracellular transfer are still to be elucidated.
1.10.2. Clandioside Catabolism
The catabolism of gangliosides occurs through the action of specific lysosomal hydrolases. In
a stepwise manner they remove individual sugar residues from the non-reducing end of the
oligosaccharide (Fig. 1.6.).
Whereas lysosomal exohydrolases directly attack membrane bound GSLs with hydrophilic
head groups extending far enough into the aqueous space, they need the assistance of srnaIl
glycoprotein cofactors. the " sphingolipid activator proteins " (SAPS) to degrade GSLs with short
oligosaccharide chahs made of 3 or less monosaccharide residues (Van Echten and Sandhoff 1993).
It is not yet clear how gangliosides are uansponed to the lysosomes for degradation. It is
generally assumed that components of plasma membrane reach the lysosomes mainly by an
endocytotic membrane flow dong the early and latc endosomal compartments. This mode1 proposes
that parts of the endosomal membranes. possibly those ennched in plasma membrane components.
bud off into the endosornal lumen forming intraendosomal vesicles. Those vesicles could finally be
delivered directly into the lysosol, the lumen of lysosomes. for degndation (reviewcd in (Fürst and
Sandhoff 1992)). However. it is possible that some other mechanisms could be invclved in the
glycolipid transfer to lysosomes. In vitro expenments showed that the G M ~ activator protein can
remove labeled gangliosides from one liposome and insert them to another. transfemng the most
abundant brain gangiioside, GM 1, at a preferential rate (Conzelmann e t al. 1982). Furthemore. the
fluorescent lipid probe, octadecylrhodamin B chloride. can be transferred from donor to acceptor
membranes in vitro by the activator protein (Kuwana et al. 1995). The activator retained its transport
25

ability at neutral pH in both mentioned cases. Thus, it is possible that the G M ~ activator protein could
have other extra and intracellular functions in vivo , involving its ability to transport a variety of
glycolipids.
REFERENCES
Agamanolis DP (1995) The Pathology of Lysosomal Storage Diseases. Pathol. Annu. 30:247-285
Banerjee A, Burg J, Conzelmann E, Carroll M. Sandhoff K (1984) Enzyme-linked immunosorbent
assay for the ganglioside Gw-activator protein: screening of normal huma. tissues and body
fluids, of tissues of GM2 gangliosidosis, and for its subcellular localization. Biol. Chem.
Hoppe-Seyler 365: 347-356
Bayleran J, Hechtman P. Saray W (1984) Synthesis of 4-melhylurnbeIIifery1-hem-D-N-
acetylg~ucosanine-6-sulfate and its use in classification of GM2 gangliosidosis genotypes.
Clin. Chim. Acta- 143:73-89
Bikker H, Meyer MF, Merk AC, de Vijlder JI. Bolhuis PA (1988) XmnI RFLP at 5q 13 detected by
a 049 Xmn 1 fragment of human hexosarninidase (HEXB). Nucleic Acids Res. 16:8 198-8 198
Brown CA, Mahuran DJ (1993) fbhexosaminidase isozymes from cells CO-transfected with a and fl
cDNA consmicts: Analysis of a subunit missense mutation associated with the adult form of
Tay-Sachs disease. Am. J. Hum. Genet 53:497-508
Brown CA, Neote K, Leung A, Grave1 RA, Mahuran DJ (1989) Introduction of the a subunit
mutation mociated with the B 1 variant of Tay-Sachs disease into the P subunit produces a P-
hexosaminidase B without catalytic activity. I. Biol. Chem. 2642 1705-2 17 10
Burg J, Bane jee A. Sandhoff K (1985) Molecular fonns of GM2-activaror protein: a study on its
biosynthesis in human skin fibroblasts. Biol. Chem. Hoppe-Seyler 366:887-891
Conzelmann E, Burg J, Stephan G. Sandhoff K (1982) Complexing of glycolipids and their
m s f e r between membranes by the activator protein for depdat ion of lysosomal ganglioside
GMZ Eur. J. Biochem. 123:455-464

Conzelmann E. Sandhoff K (1978) AB variant of infantile GM2 gmgliosidosis: drficiency of a
factor necessary for stimulation of hexosaminidase A-catalyzed degradation of ganglioside
GhiZ and glycolipid GA2. Roc. Natl. Acad. Sci. 753979-3983
Creek KE, Fischer D. Sly W (1983) Deterrninants in the uptake of lysosomal enzymes by cultured
fibroblasts. Methods EnzyrnoI. 98 :290-300
Croze E, Ivanov IE, Kreibich G. Adesnik M, Sabatini DD. Rosenfeld MG (1989) Endolyn-78. a
membrane glycoprotein present in morphologically diverse components of the endosomal and
lysosomal companments: Implications for lysosomal biogenesis. J. Ce11 Biol. 108: 1597- 16 13
De Gasperi R, Sosa MAG. Battistini S. Yeretsian J, Raghavan S. Zelnik N, Leshinsky E. et al
(1996) Late-onset Gm2 gangliosidosis: Ashicenazi Jewish farnily with an exon 5 mutation
(Tyr 180-->His) in the Hex A alpha-chain gene. Neurology 47547-552
Fishman PH, Pacuszka T. Orlandi PA (1993) Gangliosides as receptors for bacteriai enterotoxins.
Adv Lipid Res 25: 165- 1 87
Fürst W. Sandhoff K (1992) Activator proteins and topology of lysosomai sphingolipid catabolism.
Biochim. Biophys. Acta 1 126: 1- 16
Fürst W. Schubert J. Machleidt W. Meyer HE, Sandhoff K (1990) The complete arnino-acid
sequences of human ganglioside Gm activator protein and cerebroside sulfate activator
protein. Eur. J. Biochem. 192:7W-7 14
Glickrnan JN, Conibear E. Pearse BMF (1989) Specificity of binding of clathnn adaptors to signals
on the mannose-6-phosphatdinsulin-like growth factor II receptor. EMBO J. 8: 1041- 1047
Grave1 RA. Clarke JTR, Kaback MM. Mahuran D. Sandhoff K. Suzuki K (1995) The GW2
gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Mctabolic and
Molecular Bases of Inherited Disease. Vol. 2. McGraw-Hill. New York, pp 2839-2879
Griffiths G. Hoflack B, Simons K. Mellman 1. Kornfeld S (1988) The mannose 6-phosphate
receptor and the biogenesis of the lysosomes. Ce11 52:329-341
Hasilik A, von Figura K (198 1) Oligosaccharides in lysosomal enzymes. Distributions of high-
mannose and complex oligosaccharides in cathepsin D and phexosaminidase. Eur. J.
Biochem. 121:125-129

Hendrick JP, Hart1 F-U (1993) Molecular chaperone functions of heat-shock proteins. Annu. Rev.
Biochem. 62:449-384
Hiraiwa M. Soeda S, Kishimoto Y, O'Brien JS (1992) Binding and transport of gangliosides by
prosaposin. Proc. Nad. Acad. Sci. (USA) 89: 1 1254-1 1258
Hiraiwa M, Soeda S. Martin BM, Fluharty AL, Hirabayashi Y, O'Brien JS, Kishimoto Y (1993)
The effect of carbohydrate removal on stability and activity of saposin B. Arch Biochem
Biophys 303:326-33 1
Hoekstra D, Kok JW (1992) Trafficking of glycosphingolipids in eukaryotic cells: sorthg and
recycling of Lipids. Biochem. Biopshys. Acta 1 1 13:277-294
Hou Y, Tse R, M a h u m DJ (1996) The Direct Determination of the Substrate Specificity of the a-
Active site in Heterodimeric B-Hexosaminidase A. Biochemistry 353963-3969
Hubbes M, Callahan J, Grave1 R, Mahuran D (1989) The amino-terminal sequences in the pro-a
and -B polypeptides of hurnan lysosornal phexosaminidase A and B are retained in the mature
isozymes. FEBS. LE=. 249:3 16-320
Ikorine JV, Rat& MC, Desnick RI (1975) Characterization of hex S. the major residual P hexosaminidase activity in type O GM2 gangliosidosis (Sandhoff-Jatzkewitz disease). Am. J.
Hum. Genet, 27:639-650
Kaback MM. Nathan TJ, Greenwald S (1977) Tay-Sachs disease: heterozygote screening and
prenatal diagnosis--U.S. experience and world perspective. In: Kaback MM, Rimoin DL,
O'Brien JS (eds) Tay-Sachs Disease: Screening and Prevention. Alan R. Liss Inc.. New
York. pp 13-36
Klirna H, Tanaka A, Schnabel D, Nakano T, Schroder M, Suzuki K, Sandhoff K (1991)
Characterization of full-length cDNAs and the gene coding for the human GM2 activator
protein. FEBS. Lett. 289:260-264
Komeluk RG, Mahuran DJ, Neote K, Klavins MH, O'Dowd BF, Tropak M, Willard HF, et al
(1986) Isolation of cDNA clones coding for the alpha subunit of human phexosaminidase:
Extensive homology between the a and P subunits and studies on Tay-Sachs disease. J. Biol.
Chem. 26 l:8407-84 13

Komfeld R, Komfeld S (1985) Assembly of asparagine-Linked oligosaccharides. Ann. Rev.
Siochem 54:63 1-664
Kresse H, Fuchs W. Glossl J, Holtfrerich D, Gilberg W (198 1) Liberation of N-acetylglucosarnine-
&sulfate by human beta-N-acetyhexosamhidase A. J. Biol. Chem. 256: 12926- 12932
Kuwana T, Mullock BM, Luzio JP (1995) Identification of a lysosomal protein causing lipid
m s f e r , using a fluorescence assay designed to monitor membrane fusion between rat liver
endosornes and lysosomes. Biochem. J. 308:937-946
Kytzia H-J. Sandhoff K (1985) Evidence for two different active sites on human fi-hexosaminidase
A. J. Biol. Chem. 260:7568-7572
Lang L, Reitman M. Tang J. Roberts RM, Komfeld S (1984) Lysosomal enzyme phosphorylation:
Recognition of a protein-dependent determinant allows specific phosphorylation of
oligosaccharides present on lysosomal enzymes. J. Biol. Chem. 259: 14663- 1467 1
Ledeen RW, Yu RK (1982) Gangliosides:Structure. Isolation. and Andysis. Method. Enzymol.
83:139-191
Li S-C, Hirabayashi Y. Li Y-T (198 1) A protein activator for the enzyrnic hydrolysis of GM2
ganglioside. J. B iol. C hem. 256:6234-6240
Mahuran O, Novak A. Lowden JA (1985) The lysosomal hexosaminidase isozymes. Isozyrnes.
Curr. Top. Biol. Med. Res. 12:229-288
Mahuran DJ (1990) Characterization of hurnan placental ~hexosarninidase I+ - Proteolytic
processing intermediates of hexosaminidase A. J. Biol. Chem. 2656794-6799
Mahuran DJ, Neote K. Klavins MH. Leung A. Grave1 RA (1988) Proteolytic processing of human
pro-B hexosarninidase: Identification of the intemal site of hydrolysis that produces the
nonidentical Ba and Pb polypeptides in the mature P-subunit J. Biol. Chem. 263:46 12-46 18
Meier EM, Schwarzmann G. Fürst W, Sandhoff K (199 1) The human GMZ activator protein. A
subsuate specific cofactor of ~hexosaminidase A. J. Biol. Chem. 266: 1879- 1887
Nagarajan S. Chen HC. Li SC, Li YT. Lockyer JM (1992) Evidence for two cDNA clones
encoding human GM2-activator protein. Biochem. J. 282:807-8 13

Nakai H, B yers MG, Nowak NJ, Shows TB (199 1 ) Assignment of beta-hexosarninidase A alpha-
subunit to human chromosomal region 1 SqZ3--zq24. Cytogenet Ce11 Genet 56: 164- 164
Neote K, Bapat B. Dumbrille-Ross A, Troxel C. Schuster SM, Mahuran DJ, Gravel RA (1988)
Characterization of the human HEXB gene encoding lysosomal phexosaminidase. Genomics
3:279-286
O'Brien JS, Carson GS, Seo HC, Hiraiwa M. Kishimoto Y (1994) Identification of prosaposin as a
neurotrophic factor. Proc. Nad. Acad. Sci. (U.S.A.) 9 1:9593-9596
O'Dowd BF, Cumming D. Gravel RA, Mahuran D (1988) Isolation and characterization of the
major glycopeptides from human p hexosaminidase: Their localization wirhin the deduced
primary structure of the mature u and P polypeptide chahs. Biochemistry 2752 16-5226
Pearse BMF, Robinson MS (1990) Clathrin. adaptors. and sorting. Ann. Rev. Ce11 Biol. 6: 15 1- 17 1
Proia RL ( 1988) Gene encoding the human &hexosaminidase pchain: Extensive homology of
intron placement in the a- and pgenes. Proc. Natl. Acad. Sci. (USA) 85: 1883- 1887
Proia FU. Soravia E (1987) Organization of the p n e encoding the human ~hexosaminidase a
chah. J. Biol. Chem. 2625677-568 1
Sandhoff K, Conzelmann E. Neufeld EF. Kaback MM. Suzuki K (1989) The GM2 gangliosidoses.
In: Scnver CV, Beaudet AL, Sly WS, Valle D (eds) The Metabolic Basis of Inherited Disease.
Vol. 2. McGraw-Hill, New York. pp 1807- 1839
Sandhoff K, Harzer K. Fürst W (1995) Sphingolipid activator proteins. In: Scnver CR. Beaudet
AL, Sly WS. Valle D (eds) The Metabolic Basis of Inherited Disease. Vol. 2. McGraw-Hill,
New York, pp 2427-2441
Sariola H, Terava H. Rapola J. Saainen U ( 199 1 ) Cell-Surface Ganglioside GD2 in the
Immunohistochemical Deteetion and Differential Diagnosis of Neuroblastoma Am. J. Clin.
PathoI. 96:248-252
Schepers U, Glombitza G. Hoffmann A, Chabas A, O m d P. Sandhoff K (1996) Molecular
analysis of a GM2-activator deficiency in two patients with Gm-gangliosidosis AB variant
Am. J. Hum. Genet. 59: 1048- 1 O56

Schroder M, Klima H, Nakano T, Kwon H. Quintern LE, Gartner S, Suzuki K, et al (1989)
Isolation of a cDNA encoding the human GM2 activator protein. FEBS. Lett 251: 197-200
Schroder M. Schnabel D. Hunvitz R, Young E. Suzuki K. Sandhoff K (1993) Molecular genetics
of GM2-gangliosidosis AB variant: A novel mutation and expression in BHK cells. Hum.
Genet. 92:437-440
Siegel DA, W a e y SU (1994) Growth of ectopic dendrites on cortical pyramidal neurons in
neuronal storage diseases correlates with abnormal accumulation of GM2 ganglioside. J.
Neurochem. 62: 1852- 1862
Srivastava SK. Beutler E (1973) Hexosaminidase A and hexosaminidase B: studies in Tay-Sachs
and Sandhoff s disease. Nature. 24 1 :463-463
Stirling J. Leung A. Grave1 RA. Mahuran DJ (1988) Localization of the Pro-Sequence within the
Total Deduced Primary Structure of Human PHexosaminidase B. FEBS. LEïT. 23 1 :47-50
Swallow DM. Islam 1. Fox MF. Povey S. Klima H. Schepers U. Sandhoff K (1993) Regional
localization of the gene coding for the GM2 activator protein (GMZA) to chromosome 5q32-
33 and confirmation of the assignrnent of GM2AP to chromosome 3, Am, Hum. Genet.
57: 187- 193
Tse R, Wu YJ. Vavougios G. Hou Y. Hinek A, Mahuran DJ (1996) Identifkation of Functional
Domains within the a and p Subunits of P-Hexosarninidase A Through the Expression of a-(3
Fusion Proteins. Biochemisüy 35: 10894- 10903
Valentino L. Moss T, Olson E. Wang H-J. Elashoff R. Ladish S (1990) Shed Tumor Gangliosides
and Progression of Human Neuroblastoma Blood 75: 1564- 1567
Van Echten G. Sandhoff K (1993) Ganglioside metabolism. Enzyrnology. topology. and
regulation. J Bi01 Chem 268:SM 1-5344
Varki AP, Reitman ML. Kornfeld S (198 1) Identification of a variant of mucolipidosis 3 (pseudo
Hurler polydystrophy): A catalytically active N-acetylglucosarninylphosphotmsferase that
fails to phosphorylate lysosornal enzymes. Proc. Natl. Acad. Sci. (USA) 78:7773-7777
Wiesmann U, Vassella F. Hershkowitz N (197 1) Icell disease: Lakage of lysosornal enzymes
into extracellular fluids. N. Engl. J. Med. 285: lO90-109 1

Wu W. Lockyer JM, Sugiyarna E. Pavlova NV, Li YT. Li SC (1994) Expression and specificity
of human GM2 activator protein. J. Biol. Chem. 269: 16276-16283
Wu W. Sonnino S. Li YT, Li SC (1996) Characterization of an altematively spliced GM2 activator
protein, GM2A protein - An activator protein which stimulates the enzymatic hydrolysis of N-
acetylneuraminic acid. but not N-acetylgalactosamine. from GM2. J. Biol. Chem. 27 1 : 106 1 1-
10615
Xie B. Kennedy JL, M c I ~ e s B. Auger D, Mahuran D (1992) Identification of a processed
pseudogene related to the functional gene encoding the Gm activator protein: Localization of
the pseudogene to human chromosome 3 and the functional gene to hurnan chromosome 5.
Genomics 14:796-798
Xie B. McInnes B. Neote K. Lamhonwah A-M. Mahuran D (1991) Isolation and expression of a
full-Iength cDNA encoding the human GM2 activator pmtein. Biochem. Biophys. Res.
Comrn, 177:1217-1223
Zeller CB, Marchase RB (1992) Gangliosides as modulators of ce11 function. Am. .J. Physiol. Cell
Physiol. 262:C 134 1 -C 1 355

C H A P T E R II
DEVELOPMENT OF THE FLUORESCENCE DEQUENCHING
ASSAY FOR STRUCTURE 1 FUNCTION STUDIES OF THE GM2
ACTIVATOR PROTEIN

2.1 INTRODUCTION
The G m activator protein is a substrate specific cofactor for degradation of GMZ ganglioside
by lysosomal B-hexosaminidase A. It solubilizes individual molecules of the ganglioside by
interacting with both its hydrophilic oligosaccharide and hydrophobic ceramide moieties. However,
the specificity of binding is pnmarily deterrnined by the oligosaccharide moiety of the glycolipid.
From the deduced primary structure. the activator protein is predicted to contain as many as three a
helices, frorn which two are predicted to have a significant amphiphilic character. These functions
may be involved in the formation of a lipid binding cavity (Fürst et al. 1990). In vitro. the activator
can also act as a general sphingolipid transport protein (Conzelrnann et ai. 1982).
The formation of the G M ~ activator protein : glycolipid complex has been investigated by a
variety of techniques (gel filtration. electrophoresis. isoelectric focusing and ultracentrifugation)
(Conzelrnann et al. 1982). However. not only are these techniques complex and require radio-labeled
glycolipids, but none have allowed the direct kinetic measurements of glycolipid hinding. i.e. Kd and
"n". The difficulty in such measurements in part resides in the inability to immohilize gangliosides
through either their ceramide or oligosaccharide moieties without affecting activator hinding. Indirect
measurements through functional assays with Hex A have indicated a "n" value of 1 (number of
binding sites) and a Kd of 3.5 pM (reviewed in (Sandhoff et al. 1995)).
Recently. a fluorescence dequenching assay sugpstcd itself as a new method to aid in
structure / function studies of G M ~ activator protein. Originally, it was used to investigate the fusion
of sub-cellular organelles ((Hoekstra et al. 1984) and reviewed in (Hoekstra 1990; Mullock and Luzio
1992)). The principle of the assay relies upon the efficient self-quenching of the fiuorophore
rhodamine, conjugated to a saturated C 18 hydrocarbon chah (R- 18). This probe is readily inserted
into biological membranes by exogenous addition of an ethanolic solution of the fluorophore. At
concentrations up to 9 mol% with respect to total lipid, the efficiency of the self quenching is
proportional to its surface density. After membrane insertion, the localization of the probe is such that
the rhodarnine head group likely resides at the lipid-water interface, while the hydrocarbon moiety is

Figure 2.1 .:
The principIe of the fluorescence dequenching assay:
1) R- 18 (octadecylrhodamine B chloride) added to a suspension of membranes becornes
incorporated in the membranes at selfquenching concentrations.
2) The selfquenching is relieved by fusion of these membranes with unlabeled membranes.
3) Fusion results in probe dilution and increase in fluorescence which c m be rneasured.


anchored in the hydrophobic interior of the membrane. Once inserted into the membrane. the probe
does not dissociate from it by either a spontaneous transfer of free monomers through the aqueous
phase or by a collision mediated transfer process. Upon fusion of R-18 containing membranes with
membranes devoid of the probe. the surface density of the fluorophor decreases. resulting in an
increase in fluorescence that can be monitored continuously (Fig.2.1.) (reviewed in (Hoekstra 1990;
Mullock and Luzio 1992)).
Using fluorescence dequenching assay for monitoring the fusion of endosomes and
lysosomes, Kuwana et al . (Kuwana et al. 1993; Kuwana et al. 1995) isolated a protein that increased
fluorescence in their assay. It was purified and found to be 22 kDa molecule with sequence,
immunological and functional characteristics consistent with the rat homologue of human G M ~
activator protein. However, Our lahoratory showed that the human activator was not involved in
endosomal-Iysosomal fusion (Xie and Mahuran 1994). Further wark suggested that the rat protein
had leaked from the lysosome preparation (Kuwana et al. 1995). For both, the recombinant human
and rat homologue of human G M ~ activator protein, these data were consistent with the activator
acting as an R- 1 8 transfer protein.
The simplicity of the R- 18 assay and the finding that the activator c m act as R- 18 transfer
protein between egg phosphatidylcholine (PC) liposomes. as well as isolated endosomes and
lysosomes (Kuwana et al. 1995). suggested to us the possibility of using this fluorescence assay for
structure / function investigations of the G M ~ activator protein.

2.2 MATERIALS AND METHODS
R-18 was obtained from Molecular Probes. Egg phosphatidylcholine (PC) in chloroform was
from Avanti Polar Lipids. Unless otherwise stated al l other reagents were from Sigma Chemicai Co.
2.2.2. Production of the H i s a Activator Fusion Protein from E. Coli
With minor modifications we used the sarne basic methods for the synthesis by transformed
E. Coli, purification. and refolding of a Hi%-Gm activator fusion protein (Hi%-activator) as reported
by Klima et al. (Klirna et al. 1993). The method for the synthesis of the G M ~ activator fusion protein
and its vuncated form (see below) was set up by Dr.Brigitte Rigat in our laboratory. Bnefly. PCR
was used to amplify the coding sequence of the mature activator (residues 32-193) lrom pActl (Xie et
al. 199 1) in such a manner a s to allow the product to he inserted in frarne and down Stream from the
encoded N-terminal His6 in the E. Coli expression vector pQE-8 (Qiagen). A cloned insert was
confirmed by sequencing to encode the proper amino acid sequence. MRGS(H)~GSIEGR-S~~-~~~~.
In one other clone a Taq-error was identified chat produced a frame shift after codon 157 followed by
the generation of 13 new codons (MRGS(H)~GSIEGR-~32-LIST-WSCPVGSPPGTTA) before a
STOP codon was encountered. This protein (referred to as the truncated-activator) was purified and
refolded into a water soluble form in the sarne rnanner as the wild type protein.
The purifkation procedure started with the preparation of E-Coli pre-culture. One day before
main culture. 100 pl of E.Coli cells in 10 ml Super media (25 g bactotryptone, 15 g bactoyeast
extract and 5 g NaCl. per liter) were grown in the presence of ampicillin (100 pgml) and kanamycin
(50 pg/rnl), ovemight. The next day. the pre culture (IO ml) was grown in 100 ml of Super media
with ampicillin and kanamycin for lh. The expression of the cDNA insert was then induced by

adding IPTG to a fmai concentration of 3 mM. The cells were incubated for 7h and then harvested by
centrifugation (4000 g. 15 min). The pellet was kept at -70°C. ovemight
The pellet of a 100 ml culture was left on the ice for 2h to thaw and then resuspended in 10 ml
of Extraction buffer (8 M Urea 0.1 M Sodium Phosphate. 0.01 M Tris. 3% v/v f3 mercaptoethanol.
pH 8) for 2 h by gentle mixing. After extraction the ce11 debris was removed by cenuifuging (6000 g.
15 min). The crude supematant was used for further procedures.
The crude supematant was added to 5 ml of Ni-NTA resin (Quiagen Inc.. Hilden. Gemany)
and gently mixed ovemight The hexahistidine residues of the recombinant G M ~ activator protein
bound with high affinity to ~ i 2 + ions. The next day. the proteinfresin complex was loaded ont0 a
column for the washing and elution steps. The colurnn was washed with two column volumes of the
Washing buffer 1 (Extraction buffer at pH 7). and two column volumes of the Washing buffer II (6 M
Guanidine. 0.1 M Sodium Phosphate. 0.01 M Tris. 3% v/v beta mercaptoethanol, pH 7). it was then
eluted with two column volumes of the Elution buffer (Washing buffer II at pH 5). Elution fractions
were collected in different volumens. consecutively (5 ml. 20 ml, 10 ml and 15 ml). A sample of 100
pl from each fraction and each experirnental step was rnixed with 1 ml of precipitating solution (99%
ethanol, 1% acetic acid) at - 20°C ovemight and then cenuifuged and washed with 70% ethanol. To
determine the punty, as well as the molecular weight of the H i s g - G ~ 2 activator protein in sarnples
from al1 purification steps. the pellet was analyzed by SDSPAGE using the Laemrnli system
(Laemmli 1970) and a 12.5% running gel and 4.52 stacking gel. The protein was visualized by
Coomassie blue staining (Fig 2.2).
Refolding of the recombinant G M ~ activator protein was accomplished by the dilution of the
Elution buffer containing the activator protein (fraction 2 (20 ml)). with five volumes of the Folding
buffer A (2 m M reduced Glutathione, 0.2 mM oxidized Glutathione, 50 mM Tris pH 8 and 0.0038
v/v Tween 20) and stored at 4'C for 18 h. After that period, the solution was dialyzed against folding
buffer B (100 mM NaCI. 50 m M Tris pH 8 and 0.003% v/v Tween 20) for 48 h at room temperature.

Figure 2.2:
SDS-PAGE separation of the Hisfj -G~2 activator protein, in samples from different activator
protein purification steps: lysate (lane 2)- unbound fraction (lane3). wash 1 (lane 4), wash II (lane 5).
elution 1 (lane 6), elution 2 (lane 7), elution 3 (lane 8) and elution 4 (lane 9). Standard molecular
weight markers are presented in the lane 1.
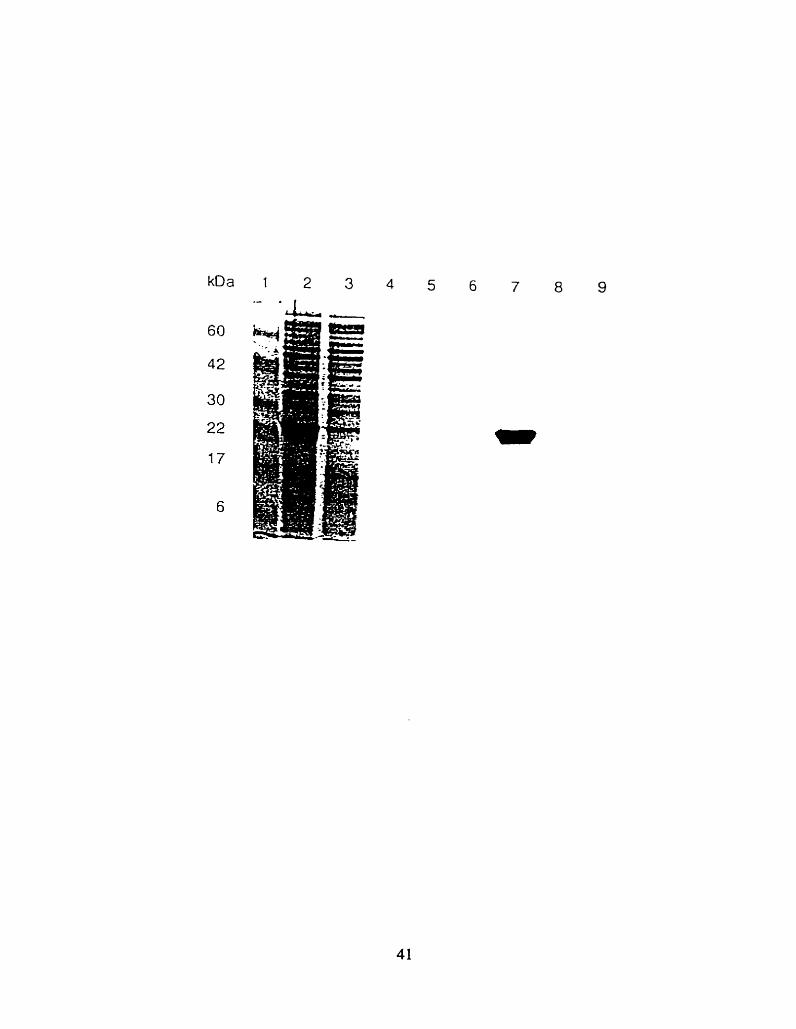

The protein was concentrated in a Stirred ce11 (Arnicon. U.S.A.) fitted with Diafio ultra filter [(YM
10.43 mm) Amicon. U.S.A.]. Concentration of the protein was carried out at CC and the protein
level was detemined by the Lowry method (Lowry et al. 195 1).
2.2.3. Pre~aration of Phos ohatidvlcholine Con_- 1 Inilamel . . lar Vesicla
Phosphatidylcholine containing large unilamellar vesicles (PC LW,). i.e. Liposomes, were
prepared with some modifications a s described by Kuwana et al. (Kuwana et al. 1995). Egg PC in
chloroform was dned under nitrogen, dehydrated under high vacuum for 1-2 h and kept at -20°C until
use. The lipid was resuspended at 5 mg/ml in NHE huffer (0.15 M NaCI. 10 mM Hepes. O. 1 mM
EDTA. pH 5) and freeze -diawed ten times to ensure entrapment of the huffer in the inner space of
vesicles fomed. The vesicles vere consecutively exmded through two polycarbonate filters (pore
diameter 200 nm and 100 nm), fitted in a mini-extruder (Avestin. Ottawa, Ont., Canada) with 19
passes (MacDonald et al. 1991). The PC-LUV, were made fresh for each experiment
2.2.4. Prepmtion of R- 18 labeled liposomes
In a total volume of 0.5 ml of NHE buffer. the PC-LUV, (0.44 pmol) were rapidly and
thoroughly mixed nt room temperature with 0.04 pmol of R- 18 (20 m M in 1 0 % cthanol). then
incubated for 15 min with gentle mixing and protected from light. Free R-IR was removed by
Sephadex G-50 gel tiltration. The 1 x 4 cm column was cluted with NHE buffer. This preparation of
R- 18 labeled liposomes was sufficient for ten assays.
2.2.5. Fluorescence de au en ch in^
Fluorescence dequenching assays were performed with some modifications as described
(Kuwana et al. 1995). In each assay, 630 nmol of unlabeled liposomes were mixed with appropriate
42

arnounts of recombinant human G M ~ activator protein with or without gangliosides. Gangliosides
were added in the 2 p l total volume of 2:l chloroform : methano1 standard solution- The assay was
initiated by the addition of 44 m o l of R- 18 labeled liposomes. The assay mixture was made up to 1
ml with SPM buffer (0.25 M Sucrose, IO m M Na phosphate buffer, 1 mM MgCl2, pH 5).
Fluorescence emission was read at 590 nm using an excitation wavelength of 560 nm. on a 650-40
fluororneter (Perkin-Eimer) in the thermostated (37') quartz cuvette.
The fluorescence dequenching of the R- 18 liposome preparations was measured and calculated
with some modifications as described by Epand et al. (Epand et al. 1995). The fluorescence intensity,
5 min after addition of R- 18 PC-LW,, was taken as Fo. At the end of the reaction, 60 pl of 20% v/v
Triton X-100 was added to the assay mixture to obtain the fluorescence value at infinite dilution of the
probe (Flm). Fluorescence was taken at any tirne point (FJ and dequenching calculated as; % R-18
dequenching = 100 (Ft-FO )/ F ~ w . The fluorescence dequenching units (Fu) were calculated by
subtraction of the baseline percentage of R- 18 fluorescence dequenching (obtained from the sample
containing al1 assay components except the activator) from each sample. Slopes. [A Fu I A T (min)].
were calculated by subtraction of the baseline dope from the dope of each sample.
2.3. RESULTS
2.3.1. Fluo rescence deauench in^ of R-18 labelcd 1'- ioosomes in the presence of recombinant
human Gm activator protein
Incubation of R- 1 8 labeled and unlabeled liposomes in the presence of 4pg of the recombinant
human activator (0.21pM), resulted in a time dependent loss of fluorescence quenching. i .e. an
increase in fluorescence over time (Fig.2.3.). Baseline fluorescence. corresponding to spontaneous
fluorescence dequenching (possibly due to dissociation from the liposomes) in R- 18 liposomes

Figure 2.3-:
Fluorescence dequenching. expressed as 5% of total (see Methods) of R- 18 labeled liposomes.
in presence of 4 pg of recombinant human G M ~ activator protein, open triangles, fit in a bat-f i t
straight iine by least squares analysis (dashed h e ) or absence of the activator (negative control. solid
squares) presented as a solid best fitted line.

2 3 4 Time (min.)

Figure 2.4a:
Fluorescence dequenching uni& Fu (see Methods), developed over 60 minutes with 1-3 pg
of recombinant human G M ~ activator protein. The best fit curves [fitted to the equation "FU=Clx T/
(C2+T)" by non-linear least squares] are shown with the actual data points.

Time (min)

Figure 2.4b.:
The time range for the linear rate of fluorescence dequenching by 1-3 pg of activator. Data
points are fitied to the bat-fit line by least squares analyses.

2 3 4
Time (min)

system without the recombinant human activator. was -2 fold lower (Fig.2.3.). After the subtraction
of the baseline slope. the resulting activator slope was 0.365. The tnincated f o m of the G M ~
activator protein (see Methods) was used as a negative control (Fig.3.2.). Unlike the full length
mature protein produced in bacteria which readily enhances Hex A's ability to hydrolyse G m (Hou et
al. 1996). the truncated protein was totally non-functional (unpublished results). In the fluorescence
dequenching assay 4pg of the tnincated activator (see Chapter III) produced a dope of 0.018. after
baseline subtraction. suggesting that it does not contain a functional hydrophobic binding site.
2.3.2. Determination of the optimal time course range for the fluorescence dequenchin~ assu
The optimal time course for the fluorescence dequenching assay was determined for three
arnounts of activator (1 pg. 2 pg. 3 pg). Fluorescence dequenching was fint measured over a 60 min
lime penod. Data from al1 samples fit the mode1 equation Fu = (C l)x (T)/ (C2+T), where C 1 and C2
are constants. with a correlation coefficient A.996. The first five minutes were required for
fluorescence stabilization; thus the fifth minute was taken a s time zero (Fig.2.4a.). The fluorescence
dequenching was linear for next 5 minutes (Fig.2.4b.).
2.3.3. Determination of the optimal G u activator protein amount for the fluorescence
gieauenchin~ assav
To determine the optimal range of activator concentrations used in the fluorescence
dequenching assay. seven levels of the activator (0.75 pg- 12 pgl ml) were tested. Slopes from each
sample were calculated and plotted against the amount of activator added (Fig.23.) . The best
concentration range for the fluorescence dequenching assay was from 0.75 pg/ ml to 3 pg/ ml
(FigSb.); however linear results werc obtained for up to 5 pg./ ml (Fig.2.5a.).

Figure 2%:
The slope of fluorescence dequenching (see Methods), versus amount of activator presenf
(pg), over an extended range of activator concentrations with data points fit to the equation
"AFu/AT=C l x pg activatod (C2yrg ac tivator)" b y non-linear least squares (dashed Line).
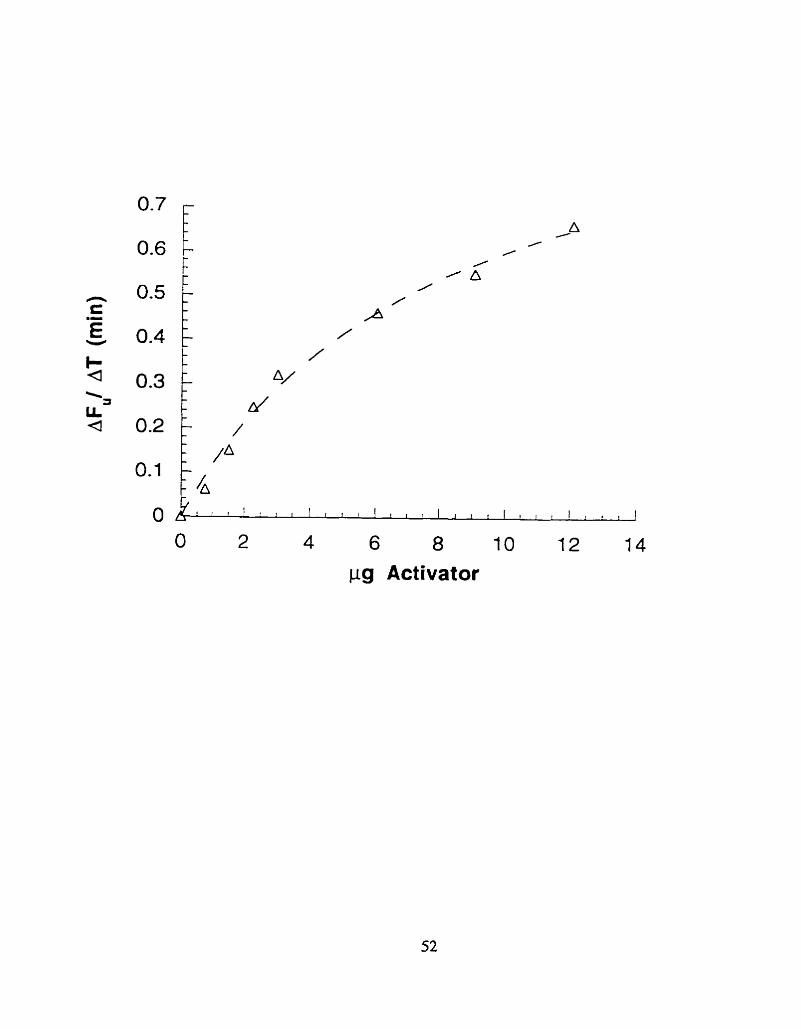
pg Activator

Figure 2.5b.:
The linear range of fluorescence dequenching (dashed he) . The amount of the activator (pg),
open triangles, in a total reac tion volume of 1 ml is plotted versus the dope of fluorescence
dequenching. open triangles, (data points are fitted to die bat-f i t line by least squares analyses).

pg Activator

Figure S.6a. :
Effect of pH on the initial rate (over 5 minutes) of fluorescence dequenching by 1-3 pg/ ml of
the activator. The dopes of the lines. i.e. (MU/ AT)/ (Apg of activator). were determined to be: pH
4= 0.052, pH 4.5= 0.095, pH 5= 0.1 16, pH 6= 0.086. pH 7= 0.050.

O 0.5 1 1.5 2 2.5 3 3.5 pg Activator

Figure 2.6b.:
The dopes (over 5 minutes) of fluorescence dequenching determined at four different
concentrations of the activator, 0.75. 1.5, 2.25, and 3.0 pg/ml. which were fitted to the best-fit line
by least squares analyses, i.e. (Mu/ AT)/ (Apg of activator), versus pH.


2.3.4. Determination of the ogtimal pH
To optimize pH for the fluorescence dequenching assay, fou. concentrations of activator (0.75
pg, 1.5 pg, 2.25 pg and 3 pg ) were analyzed at pHs from 4 - 7.5. At pH 7.5 (data not shown), 7
and 4 (Fig.2.6a) fluorescence dequenching was significantly lower than that observed at pH 4.5,5
and 6 (Fig 2.6a). The optimal pH was determined to be pH 5. At pH 5 the slope was about 18% and
27% higher than the those at pH 4.5 and pH 6, respectively; while the slopes at pH 7 and pH 4 were
reduced 50% (Fig.2-6a and Fig.2.6b.).
The data from the fluorescence dequenching assay were reproducible for each preparation of
R- 18 labeled liposomes. However, it was noted that the slope for the same sarnple size assayed under
the same conditions slowly decreased with the age of the R-18 used to prepare the liposomes. For
example, one of the experiments shown in Fig.2.4b.. produced a slope of -0.6 for 3 pg of activator
at pH 5. while an identical experiment performed latter produced a slope of -0.35 (Fig.2.5b.).
2.4. DISCUSSION
Our expenments were designed to optimize the conditions for the fluorescent dequenching
assay with the G M ~ activator protein. This assay measures the activator's ability 10 transport the self-
quenching fluorescent lipid probe octadecylrhodarnine (R- 18). between liposomes.
When labeled and unlabeled liposomes were mixed, the increase of fluorescence was observed
only in the presence of recombinant human G M ~ activator protein (tïg.2.3.). A tmncated form of the
activator, prepared from the transformed bacteria in a manner identical to the functional activator and
observed to be non-functional in the Hex A ganglioside assay, was also non-functional in the
transport of R- 18 between liposomes. These data confirm that the assay is mesuring hydrophobie
binding "activity" of the recombinant human G M ~ activator protein.

We initialiy confmed that this transfer reaction foliowed saturation kinetics, assuming that an
acidic pH of 5 would be close to the pH optimum for the reaction (Fig. 4a). The time period
correspondhg to the Linear range for the transfer was then detemined to be -5 min (Fig. 4b). This
time period was shown to be sufficient for the determination of the activator protein's transport
activity as weIl as for fluorescence dequenching comparison between different samples. The
concentration range of the activator protein producing a linear response was found to be 0.75-5 pg
with 3 pg resulting in the highest response that retained the best linearity (Fig. 5a and b).
Fmally, we confïrmed that the optimal pH was pH 5, and demonstrated that the rates of R- i 8
transport at both pH 4 and pH 7 were only about 2 fold Iess than that at pH 5 (Fig. 2.6.).
Investigating GM:! ganglioside transfer between liposomes by the activator protein. Conzelmann et
al.. (Conzelmann et al. 1982) reported that the activator c m perform such transfer at pH 7.1. retaining
15% of its pH 4.2 rate (early to laie endosomal pH is 6-5-55}. The transport of the R- 18 lipid probe
by the activator ~ q u i r e s its binding to the activator's hydrophobic binding site. while the transport of
the GMZ ganglioside requires binding to two different binding sites, Le . its hydrophilic
oligosaccharide and hydrophobic cerarnide binding sites. We propose, that both R-18 and the
ganglioside ceramide portion bind to the same hydrophobic binding site of the G M ~ activator protein
(see Results. Chapter III). As the rates of transport of R-18 at pH 4 and pH 7 differ by a smdler
amount than those of G M ~ ganglioside (Conzelmann et al. 1982). it is possible that the G M ~ activator
protein's hydrophobic binding function is not as pH sensitive as its oligosaccharide binding function.
Furthemore. as the activator. in vitro, partially retains its transport function for both R- 18 and G M ~
ganglioside at pH 7, it is possible that it can also function at neutral pH in vivo. We propose that, in
vivo, it could act as a general glycolipid sorting ancilor transport protein (see Discussion. Chapter m).
The development of the fluorescence assay for the lipid transport activity of the G M ~
activator, provides a wide spectrum of possibilities for the structure 1 function studies of the activator
protein.

REFERENCES
Conzelmann E, Burg J. Stephan G, Sandhoff K (1982) Cornplexing of glycolipids and their
transfer between membranes by the activator protein for degradation of lysosomal ganglioside
GM2. Eur. J. Biochem. 123:455-464
Epand RM, Nir S, Parolin M, Fianagan TD (1995) The role of the ganglioside GD l a as a receptor
for Sendai virus. Biochemistry 34: 1084- 1089
Fürst W. Schubert J, Machleidt W. Meyer HE, Sandhoff K (1990) The complete arnino-acid
sequences of human ganglioside Gm activator protein and cerebroside sulfate activator
protein. Eur. J. Biochem. l92:7O9-7 14
Hoekstra D (1990) Fluorescence Assays to Monitor membrane Fusion: Potential Application in
Biliary Lipid Secretion and Vesicle Interactions. Hepatology 12:6 1 S-66s
Hoeksua D, de Boer T. Klappe K, Wilschut 1 (1984) Fluorescence method for Measuimg the
Kinetics of Fusion between Biological membranes. Biochemistry 235675-568 1
Hou Y. Tse R, Mahuran DJ (1996) The Direct Determination of the Substrate Specificity of the a-
Active site in Heterdirnenc ~Hexosaminidase A. Biochemistry 353963-3969
Klima H. Klein A, Van Echten G, Schwanmann G, Suzuki K, Sandhoff K (1993) Over-
expression of a functionally active human GM2-activator protein in Eschenchia coli. Biochem.
J. 29257 1-576
Kuwana T, Mullock BM, Luzio JP ( 1993) Identification of a protein capable of causing fusion of
endosorne and lysosome membranes. Biochem. Soc. Trans. 2 1 :299-300
Kuwana T, Mullock BM, Luzio JP (1995) Identification of a lysosomal protein causing Lipid
transfer, using a fluorescence assay designed to monitor membrane fusion between rat liver
endosomes and lysosomes. Biochem. J. 308:937-946
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of
Bacteriophage T4. Nature 227:680-685
Lowry OH, Rosenbrough NJ, F m AL, Randall RJ (195 1) Protein measurement with the Folin
phenol reagent. I. Biol. Chem. 193:265-275

MacDonald RC, MacDonald RI, Menco BP, M., Takeshita K, Subbarao NK, Hu L-R (1991)
Smd-volume extrusion apparatus for preparation of large, unilamellar vesicles. B ioc him.
Biophys. Acta 106 1 :297-303
Mullock BM, Luzio JP (1992) Reconstitution of rat liver endosorne-lysosome fusion in vitro.
Methods Enzyrnol 2 19:52-6O
Sandhoff K, Harzer K. Fürst W (1995) Sphingolipid activator proteins. In: Scriver CR, Beaudet
AL, Sly WS. Vaiie D (eds) The Metabolic Bais of Inherited Disease. Vol. 2. McGraw-HiU,
New York, pp 2427-244 1
Xie B. Mahuran D (1994) The GM2 activator protein does not play a critical role in endosorne and
lysosome membrane fusion. Biochem. Biophys. Res. Commun. 20190-93
Xie B, M c I ~ e s B. Neote K, Lamhonwah A-M. Mîhuran D (199 1) Isolation and expression of a
fuil-length cDNA encoding the human GMZ activator protein. Biochem. Biophys. Res.

C H A P T E R III
EVALUATION OF THE HYDROPHOBIC AND OLIGOSACCHARIDE BINDING
FUNCTIONS AND THE IDENTIFICATION OF A SECRETORY
FORM OF THE GM2 ACTIVATOR PROTEIN

Full length cDNA clones encoding the G M ~ ac tivator protein (activator) have b e n isolated by
ourselves and others (Xie et al. 199 1 ; Nagarajan et al. 1992). The deduced activator sequence
predicts a prepro-polypeptide c h a h of 20,808 daltons (residues 1-193). a fully functional pro-
polypeptide of 18,463 daltons (residues 24- 193) and a mature chain of 17.53 1 daltons (residues 32-
193). The activator d s o has a single site for Asn-linked glycosylation. Asn63-Val-Thr. The structure
of -95% of the human GMZA gene has been charactenzed and four exons identified (Klima et al.
1991). A second altematively mRNA spliced product containing exons 1-3 and intron 3 has been
identified. Due to the presence of a STOP codon early in the retained intmn 3 sequence. the product
of the alternatively spliced mRNA is a tmncated form of the activator protein containing residues 1-
142 and an additional three residues encoded by intron 3, Val-Ser-Thr (Nagarajan et al. 1992; Wu et
al. 1996).
The only fully documented physiological role of the activator is to present the G M ~
ganglioside to the lysosomal Hex A for degradation. To do this, the activator first interacts with the
hydrophilic oligosaccharide and the hydrophobic ceramide moieties of the ganglioside molecule. with
its specificity of binding primaily determined by the former. The binding of various gangliosides to
the activator decrease in following order: G M ~ » GMI> GDla= G M ~ = GA^ (Conzdmann et al.
1982). In vitro binding studies have indicated that the terminal N-acetylgalactosamine (GalNAc) and
intemal sialic acid (NeuAc) residues of G M ~ play an important role in binding. The resulting 1 to 1
activator: ganglioside complex must then specificaily interact with Hex A (reviewed in (Sandhoff et al.
1995)). Thus. the functional assay (the enhancement of &2 hydrolysis by Hex A) for the activator is
actually a measure of at least three separate activator functions.
The above functional dornains, Le. oligosaccharide binding site(s). the hydrophobic binding
pocket and Hex A recognition site, may residc within different portions of the activator protein. It has
been suggested from studies of the GM~A protein (cornposed of residues 1- 142, above). that the
COOH teminal part of the G M ~ activator could be involved in the interaction with the GalNAc
residue of the G M ~ ganglioside andlor Hex A and that the G M ~ A protein contains the hydrophobic
64

binding pocket for the interaction with the oerarnide residue of the ganglioside (see Discussion) (Wu et
al. 19%). Previously it had b e n suggested that one or more amphiphilic a-helices of the activator
protein may form a hydrophobie binding pocket for the ceramide moiety (Fürst et ai. 1990).
The in vivo mechanisms for the intracellular transport of gang liosides are unclear. In vitro,
the activator can remove labeled gangliosides from one liposome and insert them to another. The rate
of transfer was indirectly detexmined to be -p -> GD~$> G M ~ = GA^. Thus. one of the most
abundant brain gangliosides. GM~. is transferred at a preferential rate. Furthemore, at pH 7.1, the
activator retains 15% of its uansport activity observed at pH 4.2 (Conzelmann et al. 1982). Thus. it
is possible that the activator functions as a general sorting / transport protein For gangliosides in vivo.
Prosaposin was also suggested to have this function (Hiraiwa et al. 1992). However. the G M ~
activator transfers GM 1 at 145-fold the rate of prosaposin (reviewed in (Fürst and Sandhoff 1992)).
suggesting itself as a better candidate for this function than prosaposin.
The progress in understanding the mechanism of action of the G M ~ activator protein was
harnpered by the difficulty in isolating sufficient quantities of the activator protein from human
tissues. The availability of large quantities of recombinant activator has overcome this obstacle. To
date, recombinant G M ~ activator has been produced using different expression systems. It was
synthesized in transfected COS-1 cells using a pSVL-B fusion vector (Xie et al. 1991). and in
transformed E-Coli using pQE-9 hexahistidine (Klima et al. 1993) and pT7-7 systerns (Wu et al.
1994). Methods for the expression and purification of the activator protein, that have been set up in
our laboratory use both transfected CHO cells and transformed E.Coli. Both the pFLAG-1
(unpublished data) and pQE-8 expression vectors (originally developed by (Klima et al. 1993)) have
k e n used for the latter. We found that the bacterial activator is less efficient at assisting Hex A in its
hydrolysis of G M ~ ganglioside than one produced in transfected mammalian cells, but its high yield
and ease of purification overcorne its slightly reduced specific activity. Properties of three different
GM2 activator protein expression systems in E.Cofi . are summarized in Table 3.1.

Table 3-1,:
Methods for the production of the recombinant G M ~ activator protein in E.Coii
1 Expression 1 pQE-sa
purification colurm
1 Refoiding 1 ves
wash inclusion bodies G75
a. Contains a cDNA insert encoding the human mature G M ~ activator protein. with a
hexahistidine rnoiety and a Factor Xa cleavage site insertcd at its amino terminus (Klima et al. 1993).
chromatograph y yes 20-30
b. Contains a cDNA insert encoding the human mature activator protein with an additional
(amino terminal), 9 non specfic amino acids encoded by the plasrnid (Wu et al. 1994).
no 0.1-0.2
c. Contains a cDNA insert encoding the precursor of the human G M ~ activator protein with an
amino termina. extension that encodes the Omp A signal peptide and the FLAG peptide (antibody
binding site) (data from our laboratory).
d. Predicted yield from IL of bacterial culture.

The deficiency of the G M ~ activator results in an accumulation of G M ~ ganglioside in
lysosomes, producing the phenotype of the AB variant form of gangliosidosis. To date. four
mutations of the GM2A gene encoding the activator have been linked with this disease (Xie et al.
1992; Schroder et al. 1993; Schepers e t al. 1996). The activator with the C ~ S ~ ~ ~ A I - ~ substitution was
produced in Our laboratory from both transfected COS cells and E.Coli (pFLAG-1 and pQE-8
systerns). A cDNA construct containing the $lk transition ( ~ ~ s ~ 3 8 A r ~ ) . caused ttansfected COS-
1 cells to vanscribe high levels of activator's rnRNA, but cell lysates contained only low levels of the
pro-activator protein which failed to signifcantly enhance Hex A activity (Xie et al.. 1992). However.
when sufficient mutant activator produced in E.Coli was added to the assay a significant enhancement
of Hex A activity towards G M ~ ganglioside was observed (- 1% of normal).
The development of the fluorescence dequenching assay (Chapter II) provides us with a
means of assessing the stmcture / function relationships in the G M ~ activator protein. Firstly. the
finding that the activator can act as an R- 18 transfer protein between egg phosphatidylcholine (PC)
liposomes. as well as isolated endosornes and lysosomes (Kuwana et al. 1995) d low us to use the
fluorescence assay to test for the presence of the functional hydrophobic binding site in the G M ~
activator protein. Furthemore, various glycolipids inhibit R- 18 transport by the activator, i.e. R- 18
and ceramide are bound by the same site. and their degree of inhibition c m be used to assess the
activator's additional binding affinity for oiigosaccharide residues presenr on the glycolipids.
3.2. MATERIALS AND METHODS
R- 1 8 was from Molecular Probes. Egg phosphatidylcholine (PC) in chloro f o m was obtained
from Avanti Polar Lipids. Al1 endoglycosidases were obtained from B o e h ~ g e r Mannheim. Unless
othewise stated al1 other reagents were from Sigma Chernicd Co.
67

3.2.2.Ex~ression and Purification of Hi
Spthesis by E. Coli , purification. and refolding of the Hi%-G~yrz activator fusion protein
(MRGS(H)~GSIEGR-~32-1193)- and its mncated form (MRGS(H)~GSIEGR-s~~-L l57-
WSCPVGSPPGTTA) have been described in Chapter II.
The synthesis of the G M ~ activator proteins with a nonsense mutation at codons for either
cys183 or ~ ~ ~ 1 8 5 , was accomplished by Dr.Rigat in Our laboratory. The synthesis. purification and
refolding of the mutant proteins (Act 138 and Act 185) were perfonned following the same protocol
as described for the wild type H i s g - G ~ 2 activator protein (Chapter Il).
3.2.3 .The Expression and Purification of the -6-Activator conta in in^ a cyS1 38&
Subshniéo . .
n
A cDNA construct encoding a missense mutation associated with the AB variant f o m of G M ~
gangliosidosis ( cys13*~rg) , was originally constructed by Dr.Bei Xie (Xie et al. 1992). Later,
Dr.Ftigat in our laboratory, subcloned the cDNA into the Hisg EsColi expression vector. pQE-8
(Hisg-AB). Bnefly. Hisg-AB variant activator protein was produced using PCR to amplify the
coding sequence of the mature mutant activator protein from the pAct 1 M (pSVL expression vector
containing the activator cDNA with the T~~ 2~ transition) (Xie et al. 1992). in such a manner as to
allow the product to be inserted in the E-Coli expression vector pQE-8 (see Chapter II). The
purification of the AB variant activator protein from the host E-Coli MWpREP4 (Klima et al. 1993),
transfomed with the Ligation product and grown as a bulk culture from individual colonies. was done
as described for the wild type w s 6 - G ~ ~ activator protein in Chapter II.

3.2 4. Fluorescence Deuuenching Assg
Preparation of the phosphatidylcholine-containing unilamellar vesicles and R- 18 labeled
Liposomes were done as described in Chapter II.
Fluorescence dequenching assay was performed using standard arnounts of labeled and
unlabeled liposomes in 1 ml of SPM buffer ( s e Chapter II). with or without (baseline fluorescence) 4
pg (0.2 1 nmol. 0.2 1 CIM) of H i s g - G ~ 2 activator protein, AB variant activator or activator constmcts
with nonsense mutations at codons 183 and 185, respectively and the 4 pg of tmncated activator
(0.22 nmol. 0.22 PM). For the inhibition of fluorescence. 2 pg (1.3 nmol. 1.3 /LM) of G M ~
ganglioside was added to the liposome suspension in the presence or absence (baseline fluorescence +
G M ~ ) of various forms of the activator protein. respectively. For testing the activator's binding
affinity for giycolipids 2 pg (-1-2 nmol. 1-2 PM) of various ligands were added to the liposome
mixture in the presence (4 pg, 0.21 nmol. 0.21 PM) or absence of H i s g - G ~ 2 activator protein.
Fluorescence dequenching was measured and calculated as descnbed in Chapter II.
3.2.5 Analvsis of the Distribution of Activator Moiecules with a Hiph Mannose or a Cornplex
Oliggsaccharide Moiety
The analyses of the distribution of the oligosaccharides on the semi-punfied activator from
human fibroblasts were done by Ms. Amy Leung in Our laboratory. Her data are included in this
chapter for completeness.
3.2.5.1. isolation of a Semi-Purified Sample of Newly Synthesbed Activator from N o m 1 Human
Fibroblasts
Normal human fibroblasts were grown in aMEM (minimum essential medium. Gibco-BRL)
containhg 10% FCS and antibioûcs (24X Pl50 dishes) until they reached confluency. Then 10 m M

m l was added to the medium. After two days the medium was replaced by CHO SFM II (senun
free medium, Gibco-BRL) medium plus 10 m M W C I . The cells were grown. the medium coliected
and replaced at day 7 and 12 of culture. The pooled media (-IL) was cleared (cenuifuged) and
applied to a 4.5 ml Octyl Sepharose (Pharmacia) column. The colurnn was washed wilh 200 ml of 10
rnM Na phosphate buffer pH 6 containing 0.1M NaCl and eluted in the same buffer containing 0.75%
octylglucoside (1 column volume was passed through and column elution stopped for at least four
hours, then the elution was continued). The pooled fractions containing semi-purified activator
protein (ODao, total yield 1.3 mg) were concentrated to 200 pl in a Centricon- 10 (Amicon).
3.2.5.2. Glyosidase Digestion
The effects on the apparent Mr of the activator after treatment with each of three glycosidases
dissolved in 10 mM sodium phosphate buffer pH 5 were determined. The enzymes used were; a) N-
Glycosidase F. b) Endoglycosidase F (N-Glycosidase F-Free), c) Endoglycosidase H. Each
digestion was conducted in duplicate as followed. First. the denaturation of 2.5 pl (16 pg) of semi-
purified G M ~ activator (above) was accomplished by addition of 5 pl of denatunng buffer (1.2%
SDS. 3% pmercaptoethanol. 128 octyl-bglycoside). Depending on the enzyme used ("a-c" above),
the denatunng buffer was either a) not diluted, b) diluted 1:2. or c) diluted 1:4 wirh 10 m M sodium
phosphate buffer pH 5. Then the samples were heated at 600C for 20 min. and an additional 8 pl of
phosphate buffer was added. Second. the appropnate amount of each glycosidase a) 0.4 Units in 12
pl, b) 0.2 Units in 10 pl or c) 0.004 Units in 10 pl was added to the cooled samples. Third. to one of
each duplicate leupeptin. lpg/pl of reaction mix was added. Finally al1 samples were incubated
ovemight at 37OC . Each digestion was then analyzed by Western blot (Xie and Mahuran 1994).

3.2.5.3. Western Biot Analysis
AU sarnples were individually mixed with sample buffer containing 3% SDS and 25rnM DTï
and were boiled for 1 min. The proteins in each sample were separated by SDS-PAGE using the
Laemmli gel systern (12.5% gel) (Laemmli 1970). The proteins were transferred to nitrocellulose
overnight (Brown et aL.1989). Nitrocellulose was blocked by exposure to 5% skim milk in
BLO'ITO (10 m M of Tris base, 150 mM of NaCl. 0.05% of Tween 20, pH 7.5) for 4 h, with gentle
shaking and then was incubated ovemight with 1: MO dilution (1% skim m i k in BLOïTO) of rabbit
anti-glutathione-S-transferase i G M ~ activator fusion protein antiserum. Nitrocellulose was washed 4
x 30 min. with 1% skim milk in BLOTTO and was incubated with a 1: I0.000 dilution (1% skim milk
in BLOTTO) of horseradish peroxidase-conjugated goat anti-rabbit IgG for 1 h. The nitrocellulose
was then washed 4 x 15 min. with 1% skim milk in BLOTTO, quickly rinsed with BLOTTO (no
skim milk). and dried between filter papers. It was then incubated in equal volumes of Detection
reagent 1 and Detection reagent 2 (Amersham ECL system) for precisely 1 min.. dried briefly on filter
papers. covered with Saran wrap. and exposed to Hypedilm-ECL (Arnersham) for 1 min.
3.3.1. Determinat . . . . ion of the Rec an G m Activator Prote n Binding Affinitv for
Various Glycolinids and Gan~liosides
Lf the site for the R-18 binding is the same site used by the activator to bind ceramide,
glycolipids should inhibit its rate of R-18 transfer. Furthemore the degree of inhibition should be
directly related to the strength of binding as determined by the oligosaccharide structure of the
glycolipid. When varying amounts of G M ~ ganglioside were added to the fluorescence dequenching
assay containing the near maximum level activator protein (4 pg, Fig.2.5a), the rate of fluorescence
dequenching (AF, / AT) was decreased in a dose dependent manner (Fig.3.1.). In order to compare t

Figure 3.1.:
The initiai rate (over 5 minutes) of fluorescence dequenching caused by 4 pg of activator in the
presence of hcreasing amounts of G M ~ ganghoside (solid squares) fitted to a best-fit straight line by
leas t squares analyses (solid line).

2 2.5 3 3.5 4 pg GU, Ganglioside

Table 3.2-:
Inhibition of the activator (0.2 lw-ass i s ted transfer of R- 18 by 1.3 p M of various
glycolipids. assessed b y fluorescence dequenc hing .
Glycolipid Oligosaccharide Structure % Inhibition
G ~ 2 GdNAcp( 1 -4)Galp(l-4)Glc- 3 fi1
GM3 Gal[a(2-3)NeuAc- JB(1-4)Glc- 17 f4
Gb4 GalNAcB( 1 -4)Gala(l-4)Galp(1-4)Glc- 24 M
1. Standard deviation of the slope. calculated from least squares analysis for the best fit line.

Figure 3.2.:
Fluorescence dequenching expressed as % of total (see Methods). of R- 18 labeled liposomes
in presence of 4 pg of recombinant human G M ~ activator protein, open triangles. fit to a best-fit
straight line by least squares analysis (dashed lane), 4 pg of truncated activator. inverted open
triangles, (dotted bat-fit straight Line) and a negative control. solid squares (solid bat-fit straight
he).

// e( /
/ A
/
A' /
/
A/ /
b* '- - I I I 1
I I
O 1 2 3 4 5 Time (min.)

Figure 3.3.:
Fluorescence dequenching expressed as 96 of total (see Methods). of R- 18 labeled liposomes
in presence of 4pg of recombinant human Ghiy activator protein, open triangles, fit to a bat-f it
straight line by les t squares analysis (dashed iine), 4 pg of Act 183, open squares. (dotted best-fit
straight line) and a negative conuol. solid squares, (solid best-fit straight lane).

2 3 4 Time (min.)

Figure 3.4.:
Fluorescence dequenching expressed as 8 of total (see Methods), of R- 18 labeled Liposomes
in presence of 4 pg of recombinant human G M ~ activator protein, open triangles, fitted to a best-fit
straight Iine by Ieast squares analysis (dashed line), 4 pg of Act 185, open crossed squares, (dotted
best fit straight line) and negative conuol, solid squares. (solid best fit straight line).

2 3 4 Time (min.)

he influence of different monosido, trisialo and asialo glycolipids on the fluorescence dequenching of
R- 18 over tirne, a constant amount of each glycolipid, 1.3 nmol (1.3 pM, -2 pg), was added to 0.2 1
nrnol(0.21 pM, 4 pg) of the activator. The least fluorescence dequenching was observed after the
addition of ganglioside; thus as expected. G M ~ was bound with the greatest affinity (Table
3.2.). Cornparing the data from dl the glycolipids tested, the binding affinity of the activator is; G m
>> G ~ l b >> 2 Gb4 2 ( 3 ~ 3 > GAZ (Table 3.1 .).
3.3.2. Anal~sis of the Hvdrophobic Function of Diffgrent Recombinant Human G m Activ;\fpr
Protein Construc&
The COOH terminal part of the G M ~ activator protein could be involved in its hydrophobic
binding function. In order to investigate this possibility, lhree different constructs of the activator
protein were exmined by the fluorescence dequenching assay. First, the truncated fonn of the
recombinant human G M ~ activator protein lacking 36 COOH terminal arnino acids and then, hvo
constnicts made by point mutations ( s e Methods). producing STOP codon at either Cysteine 183 or
Lysine 185.
When 0.22 nmol(4 pg, 0.22 pM ) of the mncated activator was added to the mixture of R-18
labeled and unlabeled liposomes, the increase of fluorescence was only 4.9% of that caused by the
wild type activator (0.2 1 nmol, 4 pg) (Fig.3.2.). The 4 pg (0.2 1 nmol, 0.2 1 PM) of Act 183 and Act
185 produced 6.3% (Fig.3.3.) and 9.04% (Fig.3.4.) of the functionality of the wild type activator
protein, respectively. All three constructs were also examined in 12.5 fold higher amounts (-2.6
nmol, 50 pg. -2.6 PM). Each produced only a -2 fold higher fluorescence dequenching slope (data
not shown). The addition of G M ~ ganglioside (1.3 nmol, 2 pg or 3.3 nmol, 5 pg, respectively) to
the liposomes mixture in the presence of each mutant constmct . did not cause inhibition of the
fluorescence dequenching (data not shown). Taken together, these results suggest that the truncated
and mutated forms of the activator protein lack a functional hydrophobic binding site.

Figure 3.5.:
Fluorescence dequenching expressed as % of total (see Methods), of R- 18 laheled liposomes
in presence of 4 pg of recombinant human G M ~ activator protein. open triangles. fitted to a best-fit
straight line by least squares analysis (dashed line), 4 pg of recombinant human GMZ activator in
presence of 2 pg of G m ganglioside, solid triangles (dotted bat-fit straight iine) and negative
conuol, solid squares, (solid bat-fit straight line).

O 1 2 3 4 5 6
Time (min.)

Figure 3.6.:
The initial rate of fluorescence dequenching (over 5 min) in presence of 4 pg of AB variant
activator protein, open circles, fitted to a bat-fit straight line by least squares analysis (dashed line), 4
pg of AB variant activator in the presence of 2 pg of G M ~ ganglioside. solid circles, (dotted best-fit
straight h e ) and negative control. solid squares. (soiid best-fit straight line)
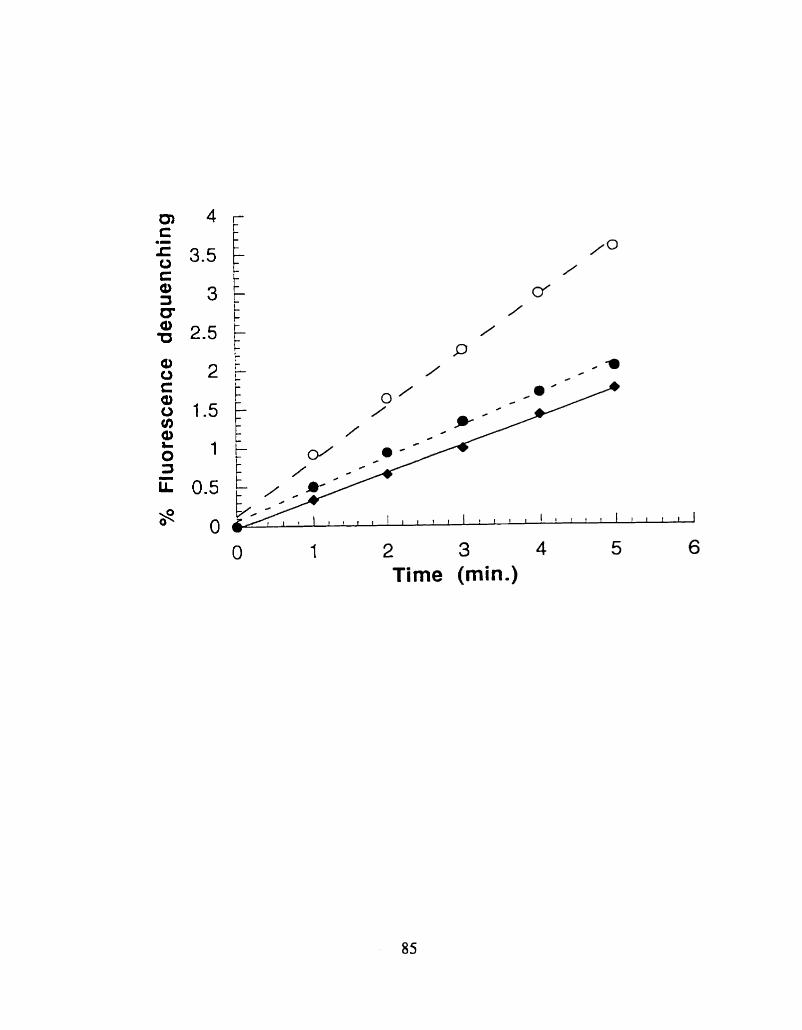
O 1 2 3 4 5 6 Time (min.)

3.3.3. Hvdroohobic Fwction of Activator Protein Con- a Point Mutation L' . . rnked to the
Ai3 Variant Forma-ned b~ w e n c e Dequenc p Assay . . .
hin
A mutant foxm of the activator protein, synthesized in transfected COS cells from the cDNA
constmct containhg the T--sC iramition in the codon for cys l38 (cys 8 3 ~ r g ) (Xie et al. 1992). as
weli as the same mutant activator produced in two E-Coli systems (using pFLAG-1 and pQE-8
expression vectors. Table 3.1.) enhance Hex A activity in the hydrolysis of G M ~ ganglioside. by
only -1% the level achieved by the wild type protein (unpublished data). Despite this fact, it is still
possible that the mutant activator has a functional hydrophobic binding site. To test this assurnption, 1
used fluorescence dequenching assay. After the addition of 0.21 nmol(4 pg, 0.21 PM) of AB variant
activator protein to the solution of R-18 labeled and unlabeled liposomes. an increase of fluorescence
was observed corresponding to 96.88 of that seen with the same amount of wild type activator
(Fig.3.5. and 3.6.). The addition of G M ~ ganglioside (1.3 nmol, 2 pg) to the mixture of liposomes
with AB variant activator (0.21 nmol, 4 pg), caused the inhibition of fluorescence dequenching of
792 (Fig.3.6.). The inhibited fluorescence was only 0.022 fluorescence units (Fu) higher than that
observed when the AB variant activator was replaced with the recombinant human G M ~ activator
protein (0.21 nmol. 4 pg) (Fig.3.5.). From the above data we can conclude that the AB variant fom
of the activator protein has a fully functional ganghoside binding site.
The location of the activator's hydrophobic binding pocket for glycolipids has not yet been
elucidated. Our working hypothesis was that the small activator protein could only contain one
hydrophobic binding site which interacts with glycolipids. gangliosides, or R- 18. This assumption
was confirmed by demonstrating that ganglioside produced a dose dependent inhibition of
fluorescence dequenching (Fig.3.1.). Furthemore, the addition to the assay of various glycolipids
produced different degrees of inhibition. These findings provided us an opportunity to directly
measure the activator's affinity for various ligands. Previously. such determinations have been made
86

indirectly, because of the need to pre-isolate the activator : ligand (radio-labeled) complex
(Conzelmann et al. 1982). Nevertheless these procedures have demonstrated the importance of the
intemal NeuAc and the terminal GalNAc residue for maximum activator binding affinity (reviewed in
(Sandhoff et al. 1995)). Our experiments confirm and extend these fîndings. As expected, we lound
that the activator binds G~v(2 ganglioside with the highest affinity nearly totally inhibiting the transfer
reaction (Table 3.2.). Interestingly. 1.3 nmo1 of G ~ l b ganglioside which has not been previously
examined, inhibited fluorescence dequenching by almost 6096. more than 2 fold the inhibition
produced by L .3 nmol of G M ~ ganglioside. These data suggest that an extra inner sialic acid may also
increase the sirength of activator's binding. Furthemore. we found that globoside. (also never
tested), and G M ~ ganglioside produce similar degrees of inhibition. This is of interest because;
whereas GA^ like GM contains a terminal GalNAc, but no NeuAc residue. GAZ was virnially non-
inhibitory in Our assay (Table 3.2.). Thus, it appears that the addition of an extra internal a-Gai
residue in increases its affinity for the activator. In sumrnary. the degrees of inhibition produced
by varioüs glycolipids and gangliosides on the R- 18 transfer assay were: %2>> G ~ l b > > G~12
G& G M ~ > GA^. The above data validate the use of the fluorescence dequenching assay for the
evaluation of the hydrophobic binding site in the activator. and the use of gangliosides andior
glycolipids inhibition of the assay to evaluate the oligosaccharide binding site(s).
The hydrophobic C-terrninals of the human and mouse G M ~ activators are highly conserved
between the two proteins. suggesting that they are cntical for activator's function (Bellachiorna et ai.
1993). To investigate the C-terminal part of the activator protein we used three different H i s 6 - G ~ 2
activator constnicts. Firstly, we exarnined its truncated form containing residues 32-157 plus an
additional 13 unrelated amino acids at its C-terminus. This construct should have similar properties to
the - 2 ~ protein produced by alternative mRNA splicing (truncated at residue 142 and containing 3
unrelated arnino acids). The G M ~ A protein. synthesized in bacteria and refolded, was reported to
retain its hydrophobic binding pocket and NeuAc recognition site based on; a) its ability to stimulate
the hydrolysis of G M ~ to GAZ by clostridial sididase. and b) its inability to stimulate the removal of
NeuAc from the oligosaccharide of the G M ~ . alone, i.e. without the ceramide moiety. in the presence
of the sarne enzyme. The G ~ A protein also Iacked the ability to stimulate the rernoval of the GalNAc
87

residue from G m by Hex A, suggesting it lacked either the GalNAc recognition site or its domain for
interacting with Hex A (Wu et al. 1996). In agreement with the latter property of the %A protein.
Our truncated protein did not enhance the hydrolysis of G M ~ by Hex A (unpublished resuits).
However, it also proved to be inactive in the fluorescent dequenching assay. indicating that the
hydmphobic binding site was either incomplete or non-functional.
Our results indicate the importance of the residues after codon 183 for the hydrophobic
binding function of the G M ~ activator protein. Both mutant f o m s of the activator, Act 183 and Act
185. were inactive in transporthg R-18 between Liposomes (Fig.3.3. and Fig.3.4.). There is only
one Cys residue lacking in the Act 183. The role of Cys residues in proteins, panicularly those
synthesized in the endoplasrnic reticulum. is often to f o m covalent intrachain disulfide bonds and
thus to stabilize the native structure of the polypeptide (Pelham 1989). The disruption of the
stabilization mechanism could result in an inability of the protein to fold properly. Interestingly. the
AB variant activator protein. with a mutation affecting Cys13? did not lack the lipid transport
activity. However, the mutant protein's ability to assist Hex A in hydrolysis of G M ~ was 75 fold less
than the normal activator's specific activity and its heat stability was dramatically reduced
(unpublished results). Thus. it is possible that the cys13*. or the novel cystine formed by it. does
not influence the conformation odand composition of the activator's hydrophobic binding site. but is
important in fonning the recognition site for Hex A.
As with prosaposin (Misasi et al. 1996; O'Brien et al. 1994) the G M ~ activator protein may
have other extracellular in vivo functions involving its ability to transport a variety of glycolipids.
The retention of this ability at neutrd pH (Fig.2.6.) which has also been previously noted by others
(Conzelmann et al. 1982: Kuwana et al. 1995). supports this hypothesis. However any extracellular
function for the activator would quire the secretion (rather than the incorporation into lysosomes) of
a significant portion of newly synthesized activator protein. The major signal for lysosomal
incorporation is one or more mannose-6-phosphate residues (M6P) linked to a high-mannose type
oligosaccharide (Kornfeld 1990). It was suggested that the activator is transported to the lysosomes
via the mannose phosphate receptors (MPRs). which recognize the M6P signal. because there was

2.5 fold increase in the amount of activator in the serum of 1-ce11 patient (Bane rjee et al. 1984).
Despite the presence of M6P signal. ceils are induced to secrete their newly synthesized lysosomal
proteins (dong with their nomally secreted proteins) when grown in the presence of NH4Cl (it
prevents the recycling of the mannose-6-phosphate receptors). Fibroblasts grown in the presence of
NH4Cl increased their secretion of the newly formed activator protein by only 2.5 fold (Burg et al.
1985). While supporthg the assumption of the ac tivator 's MPR dependent intracellular transport, the
fact that the other MPR-targeted enzymes are increased 10-20 fold under the same circumstances
(Creek et al. 1983) suggest that other pathways for lysosomal targeting may exist. We grew nomal
human fibroblasts in serum-free media containing 10 m M NH4Cl. and concentrated and semi purifted
the activator by Octyl-Sepharose chromatography (Conzelmann and Sandhoff 1979). Three sets of
duplicate samples were incubated with one of three endoglycosidases (glycopeptidase F (cleaves al1
types of Asn-linked oligosaccharides). endoglycosidase F free of any glycopeptidase F (cleaves
biantennary. but not tri- or tetraantennary complex type oligosaccharides). and endoglycosidase H
(cleaves high mannose and hybrid. but not complex type oligosaccharides)) and analyzed by Western
blotting (Fig.3.7.). The results indicate that the majonty of the activator molecules contain a complex
rather than a high mannose type oligosaccharide (Fig.3.7.). Such molecules would not contain the
mannose-6-phosphate signal necessary to divert them from the cells' secretory pathway (Kornfeld
1990). As well. a small amount of unglycosylated activator could be seen in the untreated lane;
indicating that glycosylation is not a prerequisite for the activator's transport out of the endoplasmic
reticulum (Fig.3.7.). Thus. there is a secretory form of the activator, which may have other
extracellular functions. Other experiments done in Our laboratory have identified a mannose-6-
phosphate independent pathway for the re-capture of secreted activator by cultured cells. This
pathway may explain the low level of steady state activator seen in the 1-ce11 semm and in the media of
cells grown in NH4CI. The use of the fluorescence dequenching assay with either different whole
ce11 types or membrane fractions from different tissues acting as inhibitors, may assist in identiQing
new in vivo functions for the activator.

Western blot of semi-purified activator protein from NH&-containing medium in which
normal human fibroblasts were grown. Sarnples were treated with either a) N-glycosidase F, Le.
glycopeptidase F (Glyco-F); b) endoglycosidase F. N-glgcosidase F-free (Endo-F); or c)
endoglycosidase H (Endo-H). Two samples were incubated with each glycosidase, one contained
leupeptin (+) and the other did not (-). A untreated sample is d so shown (None) dong with the
position of the glycosylated (+Oligo. Act) and unglycosylated (-Oligo. Act) forms of the activator.

Leupeptin - + - + - + O
Enzyme Giyco-F Endo-F Endo-H None _ -- .
cOligo. Act - -0ligo. A d -

REFERENCES
Bane rjee A, Burg I, Conzelmann E, Carroll M, Sandhoff K (1984) Enzyme-linked immunosorbent
assay for the ganglioside Gm-activator protein: screening of normal human tissues and body
Buids, of tissues of Gm gangliosidosis, and for its subcellular localization. Biol. Chem.
Hoppe-Seyler 365:347-356
Bellachiorna G, Stirling JL, Orlacchio A, Beccari T (1993) Cloning and sequence analysis of a
cDNA clone coding for the mouse GM2 activator protein. Biochem. J. 294:227-230
Burg J, Bane jee A, Sandhoff K (1985) Molecular forms of GM2-activator protein: a study on its
biosynthesis in human skin fibroblasts. Biol. Chem. Hoppe-Seyler 366:887-89 1
Conzelmann E Burg J. Stephan G. Sandhoff K (1982) Cornplexing of glycolipids and their
m s f e r between membranes by the activator protein for degradation of lysosomal ganglioside
GW2. Eur. I. Biochem. 123:455-464
Conzelrnann E, Sandhoff K (1979) Purification and characterization of an activator protein for the
degradation of glycolipids GM2 and GA2 by hexosaminidase A. Hoppe-Seyler's 2. Physiol.
Chem. 360:1837- 1849
Creek KE, Fischer D, Sly W (1983) Determinanis in the uptake of lysosomal enzymes by culturcd
fibroblasts. Methods Enzymol. 98:290-300
Fürst W. Sandhoff K (1992) Activator proteins and topology of lysosomal sphingolipid catabolism.
Biochim. Biophys. Acta 1 126: 1- 16
Fürst W. Schubert I. Machleidt W. Meyer HE. Sandhoff K (1990) The complete amino-acid
sequences of human ganglioside Gw activator protein and cerebroside sulfate activator
pro tein. Eur. I. Biochem. l92:7W-7 14
Hiraiwa M. Soeda S, Kishimoto Y. O'Brien JS (1992) Binding and m s p o n of gangliosides by
prosaposin. Proc. Natl. Acad. Sci. (USA) 89: 1 1254-1 1258
mima H, Klein A, Van Echten G, Schwanmann G, Suzuki K, Sandhoff K (1993) Over-
expression of a func tionall y active human GM2-ac tivator protein in Escherichia coli. B ioc hem.
J. 292:57 1-576

&a H, Tanaka A, Schnabel D, Nakano T, Schroder M, Suzuki K, Smdhoff K (1991)
Characterization of full-length cDNAs and the gene coding for the human GM2 activator
protein. FEBS. Lett. 289:260-264
Kornfeld S (1990) Lysosomal enzyme targeting. Biochem. Soc. Trans. 18:367-374
Kuwana T. Mullock BM. Luzio JP (1995) Identification of a lysosomal protein causing lipid
transfer. using a fluorescence assay designed to monitor membrane fusion between rat liver
endosornes and lysosomes. Biochem. J. 308:937-946
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of
Bacteriophage T4. Nature 227:68û-685
Misasi R, Sonce M, Carson GS, Griggi T. Lenti L. Pontien GM, O'Brien JS (1996) Prosaposin
and prosaptide, a peptide from prosaposin, induce an increase in ganglioside content on
NS20Y neuroblastoma cells. Glycoconjugate J 13: 195-202
Nagarajan S. Chen HC. Li SC. Li YT, Lockyer JM (1992) Evidence for two cDNA clones
encoding human GM2-activator protein. Biochem. J. 282:807-8 13
O'Brien JS, Carson GS, Seo HC, Hiraiwa M, Kis himoto Y ( 1 994) Identification of prosaposin as a
neurouophic factor. Proc. Nad. Acad. Sci. (U.S.A.) 9 1:9593-9596
Pelham RB (1989) Control of protein exit from the endoplasmic reticulum. Ann. Rev. Ce11 Biol.
5: 1-23
Sandhoff K, Harzer K. Fürst W (1995) Sphingolipid activator proteins. In: Scriver CR, Beaudet
AL, Sly WS, Valle D (eds) The Metabolic Basis of lnherited Disease. Vol. 2. McGraw-Hill,
New York, pp 2427-2441
Wu W, Lockyer IM. Sugiyama E, Pavlova NV, Li YT. Li SC (1994) Expression and specificity
of human GM2 activator protein. J. Biol. Chem. 269: 16276- 16283
Wu YY, Sonnino S, Li YT, Li SC (1996) Characterization of an altematively spliced GM2 activator
protein, GMZA protein - An activator protein which stimulates the enzymatic hydrolysis of N-
acetylneuraminic acid, but not N-acetylgalactosamine, from GM2. J. Biol. Chem. 27 1 : 106 1 1-
10615

Xie B, Mahuran D (1994) The GM2 activator protein does not play a critical role in endosorne and
lysosome membrane fusion. Biochem. Biophys. Res. Commun. 201:90-93
Xie B, McInnes B. Neote K. Lamhonwah A-M. Mahuran D (1991) Isolation and expression of a
full-length cDNA encoding the human GM2 activator protein. Biochern. Biophys. Ra.
Cornm. 177:1217-1223
Xie B, Wang W. Mahuran DI (1992) A C Y S ~ ~ ~ to k g substitution in the GM2 activator protein is
associated with the AB variant form of GW gangliosidosis. Am. J. Hum. Genet. 50: 1046-

C H A P T E R IV
FUTURE WORK

4.1. X-RAY CRYSTALLOGRAPHIC STUDIES OF THE G M ~ A CTIVATOR PROTEIN
The detailed three dimensional structure of the G M ~ activator protein could be estabüshed by
the x-ray crystallography. The large yield of the biologicdy functional His-6 activator protein (25-30
m a culture) and subsequent Octyl glucose purification. for obtaining only properly folded activator
protein molecules. should be of sufficient quantity and quality to altow the initial expenments on
forming activator crystals. If crystals can be formed, x-ray crystallographic studies will follow.
Furthemore, the different physical arrangement of the activator protein chains bound to the G M ~
ganglioside could be revealed by the sarne rnethod. facilitating the determination of the hydrophobic
and oligosaccharide binding sites of the G M ~ activator protein.
4.2. DETERMINATION OF THE R-18 / (;ANGLIOSIDE B INDING S ITE OF THE G M 2
ACTNATOR PROTEIN BY MUTAGENESIS
Once the putative ganglioside binding dornain is identified by crystailognphy. conservathe
point mutations can be made that will change charge and hydrophobicity of various residues in the
G M ~ activator protein. to confirm the identity of the amino acid c h a h critical for the activator's
hydrophobic binding function. It is expected that the binding site of the G M ~ activator protein will
have hydrophobic characier consistent with its function in solubilizing the G M ~ molecul: (reviewed in
(Fürst and Sandhoff 1992) ). Predicted secondary structure of the human activaior protein contains
three hydrophobic a helices which have been suggested as sites for ganglioside binding (Fürst et al.
1990). Recently, the secondary stmcture predictions showed that only one of these three a helices is
conserved in both mouse and hurnan sequences, suggesting that it is critical for the activator's
function. This lies in the sequence bounded by pro79 and pro98 of the human and mouse proteins
(Beliachioma et al. 1993).
In order to identify the R-18/ganglioside binding site of the human G M ~ activator protein, the
conservative point mutations affecting the hydrophobicity of this particular a helix could be made
followed by the isolation of the mutant His-6 activators from transformed E.Coli (Chapter II).
96

Fiuorescence dequenching assay (Chapter II) and if possible x-ray crystallography analyses would
detemine if this area is involved in gangfiosid&-18 binding. If any of these residues is critical to the
domain's ability to bind R- lS/ganglioside. it could be expected that the mutant protein (s) will noi be
able to cause the increase of fluorescence in fluorescence dequenching assay but would retain its
normal folding pattern.
4.3. IDENTIFICATION OF THE HEXOSAMINIDASE A BINDING SITE OF TAE G M 2
A CTIVATOR PROTEIN THROUGH THE EXPRESSION OF THE M OUSE / H U M AN
FUSION PROTEIN
Recently. a cDNA containing the complete coding sequence for the mouse G M ~ activator
protein was cloned and sequenced. Comparison of these data with those from the human cDNA.
demonstrate a high degree of identity in the deduced protein sequence (Yarnanaka et ai. 1994). The
alignment of the mouse and human deduced amino acid sequences were shown to contain 68% of
identical residues (Bellachioma et al. 1993). The mouse G M ~ activator protein function is shown to
be the same as for the human G M ~ activator protein. thus enhancing the degradation of the G M ~
ganglioside by the mouse Hex A.
Work in Our laboratory has shown that CHO ce11 Hex A can not interact with human G M ~
activator protein. thus it is possible that mouse activator andlor Hex A may not be able to intenct with
their human counterpm. In order to identify the Hex A binding site of the G M ~ activator, different
mouse/human fusion proteins could be used. Pnor to fusion. the extent to which mouse and human
activator proteins promote ganglioside G M ~ catabolism by autologous and heterologous
hexosaminidases will be examined. These wild type activator proteins could be synthesized in
transforrned E. Colli using pQE-8 hexahistidine system (Chapter II). If either one or both activator
proteins prove to be inactive (or show low activity compared to the species specific wild type) in
assisting the G M ~ degradation by heterologous Hex A. fusion could identify the mouse/human
domain responsible for the species-specific binding. The mouse/human fusion proteins could bé

made by the PCR three primer method (Tse et al. 1996) and produced in the same manner a s the wild
type activators. The fusion proteins could be than examined for their functionality in enhancing the
degradation of G M ~ in presence of mouse or human Hex A (depending on the results from
preliminary experiments mentioned above). as well as for the R-18 transport function in the
fluorescence dequenching assay which would serve as a control for proper folding of the activator
protein. The obtained data will point to the possible activator residues involved in the Hex A and
hydrophobic binding functions.
REFERENCES
Beilachioma G, Stirling JL, Orlacchio A, Beccari T (1993) Cloning and sequence analysis of a
cDNA clone coding for the mouse GM2 activator protein. Biochem. J. 294:227-230
Fürst W. Sandhoff K (1992) Activator proteins and topology of lysosornal sphingolipid catabolism.
Biochim. Biophys. Acta 1 126: 1- 16
Fürst W. Schubert J, Machleidt W. Meyer HE, Sandhoff K (1990) The complete amino-acid
sequences of human ganglioside Gw activator protein and cerebroside sulfate activator
protein. Eur. J. Biochem. l92:709-7 14
Tse R. Wu YJ, Vavougios G, Hou Y. Hinek A, Mahuran DJ (1996) Identification of Functional
Domains wilhin the a and p Subunits of P-Hexosaminidase A Through the Expression of a-p
Fusion Pro teins. B iochemistry 35: 10894- 10903
Yarnanaka S. Johnson ON, Lyu MS. Kozak CA, Proia RL (1994) The mouse gene encoding the
GM2 activator protein (Gm2a): cDNA sequence, expression. and chromosome mapping.
Genomics X:6O 1-604

IMAGE EVALUATION TEST TARGET (QA-3)
APPLIED IMAGE. lnc - = 1653 East Main Street
O 1993. Af~@ied Image. Inc.. All Rights Reserwd


















