
16 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT · 16 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT...
40
16 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT Peter D. Thomas 16.1 Introduction The medical management of the severely head-injured patient involves manipulations of the intracranial pressure (ICP), cerebral perfusion pressure (CPP), seizure control, fluid and electrolyte therapy and nutritional support. The goal of fluid management is homeostasis, i.e. to provide appropriate parenteral and/or enteral fluid to maintain intravascular vol- ume, left ventricular filling pressure, cardiac output, blood pressure and ultimately oxygen delivery ( ˙ DO 2 ) to tissues, when normal physiological functions are often altered by surgical and traumatic stress and by anesthetic agents. A specific aim is to prevent second- ary neuronal damage due to inadequate ˙ DO 2 to the brain; this requires adequate ventilation and oxy- genation as well as cardiac output. The most obvious consequences of inappropriate fluid resuscitation are shock from insufficient volume replacement, and pulmonary edema from overhydration. Glucose, lipids, protein, vitamins and essential trace elements are provided in order to retard the ‘auto- cannibalism’ of visceral protein for gluconeogenesis. Fluid planning must be individualized and take into account the patient’s cardiac, renal, pulmonary, nutri- tional, functional and premorbid state. Fluid and electrolyte abnormalities may result from the brain injury itself, e.g. diabetes insipidus, from therapy and from other injuries. The relationship between systemic abnormalities of water, electrolytes and acid–base metabolism and the brain can be approached from two vantage points: the effects of CNS lesions on renal function, water and electrolytes; the effects of metabolic abnormalities on the CNS. 16.2 Rationale of metabolic support The outcome after a severe head injury is influenced by age, pre-accident health, the severity of the primary injury and any of six potentially preventable second- ary insults: intracranial hematoma, raised ICP, seiz- ures, meningitis, hypoxia and hypotension (Price, 1992). About 20% of patients with head injuries also suffer injury to other systems. The injured brain is highly vulnerable to hypoxia and ischemia, which may exacerbate neuronal damage by a number of mechanisms, including the release of excitotoxic neu- rotransmitters such as glutamate (Zwimpfer and Moulton, 1993). Extracranial injury that does not result in hypotension or hypoxia appears to have little influence on outcome (Jennett, Teasdale and Braak- man, 1979). Hence preventing and aggressively treat- ing hypotension and hypoxia are essential parts of the early management of all patients with head injury. Approximately 50% of patients admitted to the hospital in a coma following head injury will require craniotomy (Butterworth and De Witt, 1989). Others who do not require craniotomy may require anes- thesia and surgery for other major injuries. The anesthesia and surgery can both affect fluid and electrolyte status. Intravenous therapy is an integral part of both the initial resuscitation phase and the later maintenance or supportive phase of management. The primary aim of resuscitation is to achieve satisfactory ventilation and restore intravascular volume, cardiac output and tissue perfusion, thereby preventing the sequelae of shock, acute renal and multiple organ failure and secondary neuronal damage. The secondary aim is correction of any underlying disorder. In adults prolonged hypoten- sion is rarely due to head injury alone, unless the injury is massive and brain death is imminent, and concealed hemorrhage must be sought. In infants and young children, hypovolemic shock may result from blood loss in a large extradural hematoma, especially if there is additional bleeding under the scalp. Shoemaker has demonstrated that O 2 transport variables ( ˙ DO 2 and oxygen consumption – ˙ VO 2 ) can reliably predict outcome in postoperative surgical patients. Conversely when these variables are manipulated in a controlled Head Injury . Edited by Peter Reilly and Ross Bullock. Published in 1997 by Chapman & Hall, London. ISBN 0 412 58540 5
Transcript of 16 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT · 16 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT...

16 FLUID, ELECTROLYTE ANDMETABOLIC MANAGEMENT
Peter D. Thomas
16.1 Introduction
The medical management of the severely head-injuredpatient involves manipulations of the intracranialpressure (ICP), cerebral perfusion pressure (CPP),seizure control, fluid and electrolyte therapy andnutritional support. The goal of fluid management ishomeostasis, i.e. to provide appropriate parenteraland/or enteral fluid to maintain intravascular vol-ume, left ventricular filling pressure, cardiac output,blood pressure and ultimately oxygen delivery (DO2)to tissues, when normal physiological functions areoften altered by surgical and traumatic stress and byanesthetic agents. A specific aim is to prevent second-ary neuronal damage due to inadequate DO2 to thebrain; this requires adequate ventilation and oxy-genation as well as cardiac output. The most obviousconsequences of inappropriate fluid resuscitation areshock from insufficient volume replacement, andpulmonary edema from overhydration.
Glucose, lipids, protein, vitamins and essential traceelements are provided in order to retard the ‘auto-cannibalism’ of visceral protein for gluconeogenesis.Fluid planning must be individualized and take intoaccount the patient’s cardiac, renal, pulmonary, nutri-tional, functional and premorbid state.
Fluid and electrolyte abnormalities may result fromthe brain injury itself, e.g. diabetes insipidus, fromtherapy and from other injuries. The relationshipbetween systemic abnormalities of water, electrolytesand acid–base metabolism and the brain can beapproached from two vantage points:
� the effects of CNS lesions on renal function, waterand electrolytes;
� the effects of metabolic abnormalities on the CNS.
16.2 Rationale of metabolic support
The outcome after a severe head injury is influencedby age, pre-accident health, the severity of the primary
injury and any of six potentially preventable second-ary insults: intracranial hematoma, raised ICP, seiz-ures, meningitis, hypoxia and hypotension (Price,1992). About 20% of patients with head injuries alsosuffer injury to other systems. The injured brain ishighly vulnerable to hypoxia and ischemia, whichmay exacerbate neuronal damage by a number ofmechanisms, including the release of excitotoxic neu-rotransmitters such as glutamate (Zwimpfer andMoulton, 1993). Extracranial injury that does not resultin hypotension or hypoxia appears to have littleinfluence on outcome (Jennett, Teasdale and Braak-man, 1979). Hence preventing and aggressively treat-ing hypotension and hypoxia are essential parts of theearly management of all patients with head injury.
Approximately 50% of patients admitted to thehospital in a coma following head injury will requirecraniotomy (Butterworth and De Witt, 1989). Otherswho do not require craniotomy may require anes-thesia and surgery for other major injuries. Theanesthesia and surgery can both affect fluid andelectrolyte status.
Intravenous therapy is an integral part of both theinitial resuscitation phase and the later maintenance orsupportive phase of management. The primary aim ofresuscitation is to achieve satisfactory ventilation andrestore intravascular volume, cardiac output and tissueperfusion, thereby preventing the sequelae of shock,acute renal and multiple organ failure and secondaryneuronal damage. The secondary aim is correction ofany underlying disorder. In adults prolonged hypoten-sion is rarely due to head injury alone, unless the injuryis massive and brain death is imminent, and concealedhemorrhage must be sought. In infants and youngchildren, hypovolemic shock may result from bloodloss in a large extradural hematoma, especially if thereis additional bleeding under the scalp. Shoemaker hasdemonstrated that O2 transport variables (DO2 andoxygen consumption – VO2) can reliably predictoutcome in postoperative surgical patients. Converselywhen these variables are manipulated in a controlled
Head Injury. Edited by Peter Reilly and Ross Bullock. Published in 1997 by Chapman & Hall, London. ISBN 0 412 58540 5

294 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
prospective manner, outcome can be improved (Shoe-maker et al. 1982, 1988). The determinants of oxygendelivery are summarized in Figure 16.1. Fluid resusci-tation augments preload, increases stroke output andtotal systemic flow, thereby increasing oxygen delivery.Care must be taken to avoid hemodilution, which maysimultaneously reduce arterial oxygen content andprevent an increase in DO2 despite improvement incardiac index. Oxygen saturation should be maintainedat 90% or greater in accordance with the recommenda-tion of the American College of Chest PhysiciansConsensus Conference on Mechanical Ventilation(Slutsky, 1993).
Following resuscitation, fluid therapy aims to main-tain euvolemia, provide normal daily requirementsand replace any continuing losses. It is usuallycombined with nutritional support.
Cerebral autoregulation determines the relationshipbetween cerebral blood flow (CBF) and metabolicneeds (metabolic coupling) and systemic hemody-namics (pressure autoregulation). When metaboliccoupling is intact, cerebral oxygen delivery is deter-mined by metabolic need and is unaffected by minorchanges in blood pressure or viscosity. Both aspects ofcerebral autoregulatory function may be impairedfollowing traumatic brain injury, so that cerebraloxygen delivery is influenced by cerebral perfusionpressure (CPP) and blood viscosity (Shoemaker et al.,1988). Thus maintaining a normal CPP is a key aspectof the management of the head-injured patient.
Most severely injured patients develop only smallchanges in serum electrolyte concentrations, eventhough there are large fluid and electrolyte shiftsbetween body compartments. Thus it is important tounderstand the physiological basis of the common
electrolyte problems, the factors controlling electrolytedistribution and extracellular fluid volume and thedistribution of administered fluids since the choice ofintravenous fluids will influence brain metabolismand volume (Sutin, Ruskin and Kaufman, 1992). Fromthis knowledge appropriate therapeutic strategies canbe developed.
16.3 Basic principles
16.3.1 FLUID DISTRIBUTION
The total body water content of humans is approx-imately 60% of body weight (Figure 16.2). Two-thirds is located in the intracellular and one-third inthe extracellular compartment (Harrison, Darrowand Yannet, 1936). The extracellular compartmentcan be further subdivided into interstitial (protein-poor) and intravascular (protein-rich) compartments.Normally, 75% of the extracellular water is containedwithin the interstitium and 25% in the vasculature.There are marked differences in electrolyte contentbetween intracellular and extracellular fluids: potas-sium is predominantly intracellular, and sodium andchloride extracellular (Gamble, Ross and Tisdall,1923). The energy-consuming Na+–K+ pump is
Figure 16.1 Systemic oxygen delivery – DO2 (ml/min) – isdetermined by the product of the oxygen content of theblood and total systemic blood flow. The major determinantsof each component are pictured. Hb = hemoglobin; % SAT =percentage saturation of Hb; PaO2 = partial pressure ofoxygen in arterial blood; 0.0031 = solubility coefficient foroxygen in plasma.
Figure 16.2 Distribution of body water in a 70 kg man (45.5liters).

BASIC PRINCIPLES 295
required to maintain these concentration gradients,which are sustained during sodium overload ordepletion states. Distribution of water between theintracellular and extracellular compartments isdetermined by osmosis.
Extracellular fluid includes water ‘attached’ to boneand dense connective tissue, as well as free trans-cellular or ‘third-space’ fluid formed by secretionsfrom epithelia and other membranes. Free trans-cellular fluid includes cerebrospinal fluid (CSF), urine,gastrointestinal and other secretory fluid, which mayhave strikingly different osmolalities from the twomajor body fluid compartments (Shoemaker, 1982).Although this third space may be greatly expanded indisease states such as ascites, pleural effusion, para-lytic ileus or bowel obstruction, it is separated fromother body fluid compartments by epithelium andtherefore does not communicate freely with them. Thethird space can be thought of as ‘parasitic’ – it isgenerated at the expense of the other two compart-ments but is not available to ‘come to their rescue’ instates of hypovolemia. Since most forms of expansionof the extracellular fluid do not involve the third spacefluids and the amount of water attached to bone anddense connective tissue is relatively fixed, the normalbody water distribution can be simplified as shown inFigure 16.3.
Cell membranes are normally relatively impermea-ble to ions but freely permeable to water. Water,electrolytes and energy substrates continually move inand out of cells to maintain a dynamic steady-staterelationship and major fluid and electrolyte shiftsoccurring with disease are often associated with onlyminimal serum concentration changes. For example, adecrease in extracellular sodium concentration andosmolarity following an infusion of 5% dextrose inwater is followed by diffusion of water into the cellsuntil the osmotic pressure between the two compart-ments is equal. Conversely, an increase in extracellularsodium concentration and osmolarity after infusion ofhypertonic saline is followed by movement of waterfrom the intracellular to the extracellular compart-ment. Infusion of an isotonic electrolyte solution doesnot alter the osmolarity of the extracellular compart-ment and therefore does not lead to shifts of water intoor out from the intracellular compartment.
Water movement between the two components ofthe extravascular space is determined primarily bydifferences in protein concentration. Colloid osmoticpressure (COP) is the net osmotic pressure across thecapillary membrane resulting from the impermeabil-ity of the endothelium to plasma proteins. Becausephysical confinement of protein molecules producesan osmotic imbalance, water moves from interstitialspace into the blood stream. Crystalloid moleculescannot establish a pressure gradient because theymove freely across the capillary membrane.
Protein molecules are negatively charged andattract a small number of positive ions, preventingthem from diffusing across the endothelium. Theseretained ions produce an additional osmotic pressure(called the Donnan equilibrium effect). Some 60% ofnormal COP is produced by protein molecules (75%by albumin, the rest mainly by globulins and fibrino-gen); 40% is produced by the electrostatically heldcations. However, if the plasma protein concentrationrises, the influence of the Donnan equilibrium effecton COP increases disproportionately, producing acurvilinear relationship between total protein concen-tration and COP.
COP is measured by placing a protein solution onone side of a semipermeable membrane and a protein-free solution on the other side. Fluid moves throughthe membrane until a pressure is generated thatprevents further osmosis. This pressure is defined asthe COP (Morissette, 1977). Plasma COP in normalambulatory patients is approximately 25 mmHg, insupine patients 22 mmHg and in an Intensive CareUnit patient 18–20 mmH (Weil et al., 1974, 1981;Rackow, Fein and Leppo, 1977). Although protein isthe primary determinant of osmotic pressure inperipheral tissues, it contributes very little to the totalnumber of particles dissolved in plasma. Total plasma
Figure 16.3 Simplified distribution of body water in a 70 kgman. The forces that influence fluid distribution between thecompartments are shown in italics.

296 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
osmolality is normally 285 mosmol/kg, of whichplasma protein accounts for less than 1 mosmol/kg.
Infusing an iso-oncotic solution such as 5% albuminin isotonic saline expands the intravascular spacewithout producing a shift of water from other com-partments. A hyperoncotic solution such as 25%albumin expands blood volume to a extent greaterthan the volume infused by drawing water andelectrolytes from the interstitium. However waterdoes not diffuse from the intracellular space ifextracellular osmolarity remains unchanged.
Starling defined the forces influencing the bulkmovement of water between the vascular and inter-stitial compartments (Starling, 1896). Pappenheimerand Soto-Rivera derived a mathematical expressionfor these forces, referred to as the Starling equation oftranscapillary exchange: (Pappenheimer and Soto-Rivera, 1948).
Qf = Kf [(Pc – Pi) – � (�c – �i)]
where Qf = total fluid flow across the capillarymembrane; Kf = fluid filtration coefficient; Pc =capillary hydrostatic pressure; Pi = interstitial hydro-static pressure; � = osmotic reflection coefficient; �c =capillary oncotic pressure; �i = interstitial oncoticpressure.
The four pressures in this equation are called theStarling forces. The net driving pressure favoringfiltration is Pc – Pi. The hydrostatic pressure within thecapillary is the major force driving fluid into theinterstitium and is essentially unopposed by theinterstitial hydrostatic pressure. This is usuallyslightly negative and approaches zero or becomesslightly positive only when substantial amounts ofedema accumulate (Meyer, Meyer and Guyton, 1968;Guyton, Granger and Taylor, 1971). The plasma COPis thus the only force acting to retain fluid within theintravascular space. The interstitial COP works in theopposite direction; the net effect of these opposingforces is described by �c – �i
Kf, the filtration coefficient, has two components: Lp,the hydraulic conductivity, which describes howrapidly fluid can pass through the microvascularexchange barrier (capillary membrane, interstitial geland terminal lymphatics) and S, the capillary surfacearea available for filtration (Granger, 1979; Harms etal., 1981; Demling et al., 1982). If either component ofKf increases, e.g. through damage to the endothelialmembrane or dilatation of precapillary vasculature inresponse to increased cardiac output (CO), the rateand amount of fluid filtered increase independently ofchanges in the Starling forces (Peters and Hargens,1981).
The reflection coefficient � defines the capacity ofthe capillary membrane to prevent translocation of
proteins. If � = 1, the membrane is totally impermea-ble and proteins are able to exert their full oncoticforce; if � = 0, the membrane permits protein to passwithout impedance (Peters and Hargens, 1981). �varies in capillary membranes throughout the body,being approximately 0.9 for systemic and 0.7 for lungcapillaries (Wittmers, Bartlett and Johnson, 1976). Thenet effect of the Starling forces across capillaries is toproduce fluid movement from the intravascular to theinterstitial space. Accumulation of interstitial fluid inthe lung is much more life-threatening than peripheraledema formation and there has been great interest inevaluating factors that are likely to reduce the risk ofclinically significant pulmonary edema (Allen et al.1987; Civetta, 1979; Crandall et al., 1983).
Modest increases in pulmonary capillary hydro-static pressure or decreases in plasma oncotic pressuredo not result in pulmonary edema because the lunghas mechanisms for resisting accumulation of inter-stitial fluid. In the first place, the pulmonary inter-stitial hydrostatic pressure (Pi) is normally slightlynegative (Civetta, 1979; Levine et al., 1967; Taylor et al.,1982). When fluid first begins to accumulate in theinterstitium, the pressure rapidly increases (i.e. com-pliance is low). This increase in hydrostatic pressureopposes any further fluid entry by decreasing thehydrostatic pressure gradient, Pc – Pi (Allen et al.,1987). However, as interstitial fluid pressure risesabove atmospheric, resistance to fluid transportthrough the interstitium decreases markedly andfurther increases in fluid volume cause only minimalincreases in interstitial hydrostatic pressure (Levine etal., 1967; Goldberg, 1980). Pi appears to level offbetween 1 and 5 mmHg in severe pulmonary edema(Battacharya, Gropper and Staub, 1984).
A second mechanism which resists the accumula-tion of pulmonary interstitial fluid is a decrease in theinterstitial oncotic pressure resulting from the accu-mulation of protein-poor fluid (Granger,1979). Pulmo-nary interstitial oncotic pressure is normally about75% of the plasma level and the oncotic gradientacross the pulmonary capillary membrane (�c – �i) isonly 4–6 mmHg. If serum COP decreases, interstitialoncotic pressure will decrease proportionately as fluidenters the interstitial space, avoiding a change in theoncotic gradient (Granger, 1979; Demling, 1980; Deml-ing et al., 1979).
Thirdly, large protein molecules such as albumin arenormally excluded from a substantial part of theinterstitial fluid volume by the density of the interstitialmatrix. However, as the degree of hydration increasesthe fibers in the interstitial matrix are stretched apart,allowing protein molecules to enter previouslyunavailable space (Granger, 1979). This lowers �i,widens the transcapillary oncotic gradient (�c – �i), andthus opposes further pulmonary edema formation.

BASIC PRINCIPLES 297
Lymphatic drainage is the fourth and probablymost important safety factor. Experimental studieshave shown that the lymph flow rate can increasetenfold when interstitial fluid volume or pressureincreases.
The blood–brain barrier is formed by a closelywoven mesh of capillary cells joined by a continuousarray of tight junctions (zonula occludens) with aneffective pore size of 0.7–0.9 nm. Because the blood–brain barrier is a lipid membrane with small pores, itis easily permeated by water and very small orlipophilic molecules. The blood–brain barrier is rela-tively impermeable to electrolytes, water-soluble non-electrolytes and plasma proteins. The endothelial cellsin peripheral tissues are joined by interspersed tightjunctions and the effective endothelial pore size isabout 4–5 nm. Because of the large pore size inperipheral tissues, the endothelial membrane is freelypermeable to water and electrolytes, but only partiallypermeable to plasma proteins. In the peripheraltissues there is an active lymphatic system, whichhelps reabsorb interstitial fluid. In the brain, however,there are no lymphatics. When the blood–brain barrieris injured, the barrier becomes permeable to electro-lytes and large plasma proteins such as albumin(Zornow et al., 1988). Hypertonic fluids will onlyremove interstitial and intracellular fluid from regionsof the brain where the blood–brain barrier is intact,and will have no effect on areas where the blood–brain barrier is permeable to electrolytes.
The difference in membrane function between theblood–brain barrier and the endothelium in periph-eral tissues is of practical importance. For example, ifthe intravascular concentration of sodium is increasedfrom 140 mmol/l to 150 mmol/l, there will only be asmall and short-lasting effect on the peripheral inter-stitial tissues, owing to the free movement of bothsodium and water across the endothelium. Howeversuch a change in sodium concentration will have apronounced effect on the brain. The osmotic pressuregradient will increase drawing water from the braininterstitium to the intravascular space.
16.3.2 OSMOLALITY AND TONICITY
Osmolality is the number of dissolved particles in akilogram of solvent (water) and determines theosmotic force of the solution (Shoemaker, 1982). Cellmembranes are highly permeable to water, which willdiffuse across them rapidly to maintain osmoticequilibrium between the extracellular and intracel-lular fluid compartments (Brobeck,1973). Hence therelative volumes of the compartments will be deter-mined by their sodium and potassium content.
Plasma osmolality can be measured directly orcalculated approximately by means of the formula:
POSM = 2PNa+ + glucose + urea
where the concentration of sodium, glucose and ureaare all expressed in mmol/l (Worthley, Guerin andPain, 1987).
The plasma sodium (PNa+) level is multiplied by
two to account for anions. This rule of thumb does nottake into account the low-concentration cations suchas potassium, calcium and magnesium or organicmolecules such as creatinine, amino acids and lactate.This error of exclusion is balanced by the presence ofmultivalent anions such as sulfate and phosphatewhich contribute fewer particles per equivalent thando univalent ions.
Although urea is included in the calculation ofosmolality, it diffuses readily across cell membranesand does not influence relative compartment volumes(Brobeck,1973). Exogenous solutes such as mannitol oralcohol may create an ‘osmolar gap’ between themeasured and unmeasured osmolalities (Table 16.1).
This formula must be interpreted in conjunctionwith the clinical assessment of fluid status as, forexample, an increased osmolality may result eitherfrom excess sodium or a deficit of water.
Tonicity defines effective osmolality, i.e. the osmoticforce due to the particles which cannot permeatebetween compartments. Because all body fluids mustbe in osmotic equilibrium, changes in tonicity, regard-less of their cause, will result in fluid shifts betweenthe ECF and the ICF in order to re-establish osmoticequilibrium. The administration of hypertonic fluidssuch as 50% dextrose or hypertonic saline will increasethe tonicity of the ECF and result in fluid shifts intothe extracellular compartment. Thus patients withosmotic hypertonicity will have a contracted intra-cellular compartment.
This effect had been applied to the treatment ofpatients with cerebral edema in whom intracellularand extracellular volumes can be decreased by achiev-ing hypernatremia. Conversely in hyponatremic statesthe tonicity of the ECF is decreased and water mustshift into and expand the intracellular fluid to main-tain osmolality. As brain expansion is limited by theconfines of the cranial vault, severe hyponatremia
Table 16.1 Osmotically active particles
Extracellular fluid Intracellular fluid
Sodium PotassiumGlucose Magnesium(Mannitol) Inorganic phosphateUrea* Urea*Ethanol* Ethanol*
*These particles are distributed freely in body water. Otherparticles are unable to permeate compartments.
298 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
may result in raised intracranial pressure (ICP). Ureaincreases osmolality but not tonicity because it passesfreely through biological membranes, and will notalter the intracellular volume,.
16.3.3 DISTRIBUTION OF ADMINISTERED FLUID INTHE BODY
All infused fluids may be considered as being com-posed of plasma equivalents (colloidal solutions),saline equivalents (crystalloid solutions) or waterequivalents. Plasma equivalents remain essentiallyconfined to the circulation as a result of their oncoticpressure. Saline equivalents are distributed betweenthe interstitium and plasma, but remain restricted tothe extracellular fluid compartment because of theirsodium content. Water equivalents are distributedaccording to the distribution of water in the body(Figure 16.2).
(a) Plasma equivalents
Plasma equivalents include blood, its components andcolloids. Blood may be administered either whole, orin its component form; the latter is preferred, asfractionation optimizes the availability of the variousblood components for special situations and improvesthe survival of the stored blood elements. Its use islimited by cost and availability, the risks of allergicreactions and infection, the delays necessary for cross-matching and a limited shelf life.
Patients who are bleeding acutely are best treated inpart with whole blood since this also replaces clottingcomponents and platelets. Otherwise hypovolemiacan be treated effectively by colloid or crystalloidsolutions, and oxygen transport function can berestored by red-cell concentrates, although these takeseveral hours to be fully effective since storage impairsoxygen dissociation.
The absolute minimum acceptable hematocrit in thetrauma patient is difficult to determine. The goal ofblood transfusion is to maintain an adequate level of
tissue oxygenation, but the focus in clinical medicinehas been on the level of serum hemoglobin or thehematocrit. It has been estimated that tissue DO2 isoptimal when the hematocrit is 33% (Sutin, Ruskinand Kaufman, 1992), but the effect on DO2 may not bethe same as the effect on oxygen utilization. Aconsensus development conference on perioperativered cell transfusion concluded that available evidencedid not support the recommended practice of main-taining hemoglobin levels at 10 g/dl or hematocrits at30% (Consensus Development Conference, 1988). Inthe United States, the National Blood Resource Educa-tion Program guidelines for red blood cell transfusionstate that adequate oxygen-carrying capacity can bemet by a hemoglobin of 7 g/dl (hematocrit of approx-imately 21%) or even less when the intravascularvolume is adequate for perfusion. The guidelines addthe caveat that the decision to transfuse a givenpatient should be based on consideration of thepatient’s age, etiology and degree of anemia, hemody-namic stability and presence of coexisting cardiac,pulmonary or vascular disease. While the normalbrain is very tolerant to anemia, the limits of iso-volemic anemia are unknown. Because autoregulationmay be impaired after injury, it cannot be assumedthat the safe lower limit of the hematocrit is the sameas in the neurologically normal subject.
Colloidal solutions comprise preparations of plasmaor high-molecular-weight synthetic substances (Table16.2). These fluids do not cross capillary membranesreadily and are initially confined mainly to theintravascular space. If the colloidal solution has anoncotic pressure that is higher than that of the plasmainterstitial fluid will move into the intravascular spaceand expand the plasma volume by more than theadministered volume of colloidal solution, hence theterm ‘plasma expander’.
Plasma-protein derivatives
These include 5% normal serum albumin, whichcontains human albumin 50 g/l and sodium
Table 16.2 Colloidal solutions
TypeVolume
(ml)Mean mol.wt (kDa)
Proteincontent
Sodium content(mmol/bottle)
Oncoticpressure Other constituents
Half-life Side effects(%)*
Cost/bottle(Aust$)
Normal serumalbumin
500 69 5% (25 g) 70 Iso-oncotic – 5–10 d 0.02 (0.004) 60.00
Concentratedalbumin
100 69 20% (20 g) 10 � 5 – 5–10 d 0.01 (0.003) 85.00
Dextran 70(Macrodex)
500 70 6% 77 or 0 � 2.5 – 12 h 0.07 (0.017) 8.00
Polygeline(Haemaccel)
500 35 3.5% 72 Iso-oncotic Potassium, 5 mmol/l;calcium, 6 mmol/l
4 h 0.15 (0.05) 12.50
*Percentage incidence of all reactions. Percentage incidence of major (grade-3 and grade-4) reactions in parentheses.

BASIC PRINCIPLES 299
140 mmol/l, concentrated (20%) albumin, which con-tains 200 g/l of albumin and less sodium (100 mmol/l).The latter is usually restricted to hypoproteinemicpatients with either hypernatremia or fluid overload. A500 ml infusion of 5% albumin expands the intra-vascular volume by 450–500 ml (Lamke and Liljedahl,1976). After 2 hours, under conditions of normalcapillary permeability, 90% remains within the intra-vascular space (Rainey and English, 1988). Eventuallythe administered albumin is distributed throughout theextra cellular space (Rainey and English, 1988). Afterinfusion of 100 ml of concentrated albumin (20%), theplasma volume continues to increase over the next30–60 minutes to achieve a final blood volumeexpansion of 400–450 ml. Redistribution of 350 ml ofinterstitial fluid to the intravascular space is necessaryfor this to occur. In patients with extracellular or totalbody water depletion, equilibration is slow andincomplete (Beecher, 1945). Therefore, in acute hypovo-lemia, 5% albumin should be given rather than thehyperoncotic form. The 20% albumin solution isusually used in patients with concomitant hypovole-mia and elevated extracellular fluid volume.
The duration of vascular retention and the hemody-namic effects of infused albumin solutions dependgreatly on the patient’s disease state. This variabilitymay result from ‘leakage’ into the interstitium, prefer-ential binding of albumin in the skin and wound, orincreased catabolism.
Albumin has several unique effects that differentiateit from other colloids as well as from crystalloids(Emerson, 1989). Albumin can bind reversibly withboth anions and cations, which allows it to regulatethe extracellular concentration of various substancessuch as iron, lipids and bilirubin (Emerson, 1989).Albumin also has the ability to act as a free radicalscavenger and may limit lipid peroxidation (Holt,Ryall and Campbell, 1984; Wasil et al., 1987; Stocker,Glazer and Ames, 1987; Pirisino et al., 1988). Theseproperties may in the future become the major reasonfor choosing albumin as a resuscitative fluid.
Albumin may also play a role in maintainingnormal microvascular permeability to protein mole-cules. Endothelial cells contain pores through whichprotein molecules may leave the vascular space.Albumin may help regulate the permeability of thesepores. Hypoalbuminemia increases capillary permea-bility and restoring the albumin level reduces permea-bility to normal (Harms et al., 1981; Demling et al.,1982). However, the intravascular albumin level neces-sary to maintain normal microvascular permeability isnot known.
Plasma-protein solutions are heated to 60°C for 10hours during processing and are free of transmissiblediseases (Rainey and English, 1988). Severe side-effects, such as hypotension, which is probably due to
kinin activation, are rare. Because of their long half-lifecare must be taken to avoid circulatory overload inpatients who have been resuscitated with plasma-protein solutions and appropriate hemodynamic mon-itoring is required.
Synthetic colloidal solutions
Dextrans are polysaccharides composed of glucosemolecules polymerized into chains of various lengths.They are classified according to molecular weight andare available in isotonic saline or 5% dextrose. Becauseof their shape dextran molecules are hyperoncotic –i.e. they have the water-attracting equivalent ofseveral individual molecules; thus they produce aplasma-volume expansion of about 120% of theinfused volume (Scheinkestel et al., 1989).
Potentially life-threatening toxic effects may compli-cate dextran administration, including acute renalfailure, anaphylaxis and bleeding diathesis (Atik,1969). Dextran 40 has been associated with acute renalfailure due to irreversible plugging of the renaltubules; Dextran 70 is rarely associated with thedevelopment of acute renal failure.
The incidence of anaphylactoid reactions after dex-tran administration was reported to be 0.032%. Dextran40 produced fewer reactions than Dextran 70 and severereactions were uncommon (Ring and Messmer, 1977).
Dextran 70 and, to a lesser extent, Dextran 40produce a dose-related hemostatic defect. This has anumber of mechanisms but is due primarily to areduction in platelet adhesion and aggregation nor-mally mediated by factor VIIIR:antigen activity (Alex-ander et al., 1975). Dextran also lowers the levels of allclotting factors by hemodilution, coats blood vesselwalls and cellular elements and impairs the elasticityand tensile strength of fibrin clots (Atik, 1967; Weiss,1967; Adelson, Crosby and Roeder, 1955; Karlson et al.,1967; Muzaffar et al., 1973). Primary hemostasis isaffected and clinically the picture mimics von Will-ebrand’s disease. Bleeding is more likely in patientswith pre-existing coagulation abnormalities. To mini-mize this risk, the volume of dextran infused shouldbe limited to 20 ml/kg/d or 1.5 g/kg/d (Atik, 1967).Some 50–70% of infused dextran is excreted by thekidneys and the rest is slowly metabolized.
Because of their interference with coagulation,dextrans are rarely used in the management ofseverely injured patients.
Polygeline (Haemaccel, Hoechst) consists of urea-bridged gelatin, molecular weight range 5000–50 000;mean 35 000, derived from cattle-bone gelatin andsuspended in saline. It is an effective plasma volumeexpander in critically ill patients (Edwards et al., 1989;Mishler, 1984). The low-molecular-weight gelatin por-tion distributes readily through the ECF and produces
300 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
a plasma volume expansion of only about 70% of theinfused volume. Renal excretion accounts for 85% ofelimination, fecal excretion for 10% and the remainderis metabolized to non-essential amino acids. Polyge-line contains potassium and calcium and this must beremembered when large volumes are infused. Rapidinfusions cause histamine release, which usually takesthe form of urticaria, but anaphylaxis has beenreported (Scheinkestel et al., 1989). Gelatin productsare not associated with renal failure or coagulopathy.
(b) Crystalloid solutions
Crystalloids are isotonic mixtures of sodium chlorideand other physiologically active solutes (Table 16.3).
Sodium is the major component of crystalloid fluidsand the distribution of infused sodium will determinethe distribution of infused crystalloid fluids. Sincesodium is the major solute in the extracellular spaceand 80% is extravascular (Edelman and Leibman,1959), infused sodium will reside primarily in theextravascular compartment.
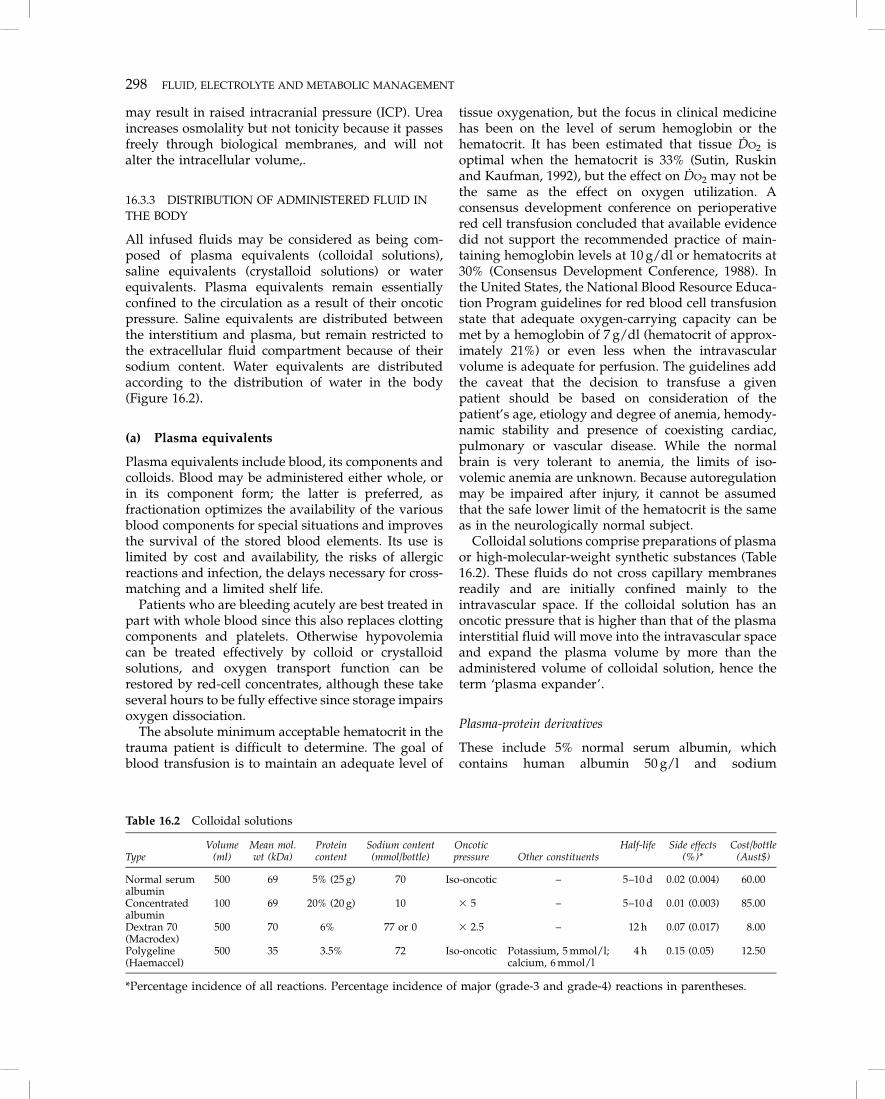
Physiological saline and Hartmann’s solution arethe two most frequently used crystalloids and theirvolume-expanding effects are identical (Cervera andMoss, 1975). Figure 16.4 shows the effect of colloidand crystalloid infusions on blood volume in criti-cally ill adults. Physiological saline and Hartmann’ssolution are both freely permeable across the vas-cular membrane and distribute evenly throughout
Table 16.3 Composition of commonly prescribed electrolyte solutions
Crystalloid solutions
Isotonic(0.9%) saline
Hartmann‘s (Ringer’slactate) solution
Water-equivalent solutions
5% dextrosein water
4% dextrosein 0.18% saline
Glucose (g) – – 50 40Calories (kcal) – – 205 164Na+ (mmol/l) 150 131 – 30K+ (mmol/l) – 5 – –Cl– (mmol/l) 150 111 – 30Ca2+ (mmol/l) – 2 – –Lactate (mmol/l) – 29 – –Osmolarity (mosmol/l) 300 279 277 282pH 6.0 5.1 3.5–6.5 3.5–6.5
Figure 16.4 The influence of colloid and crystalloid infusion on blood volume in critically ill adults. (Source: redrawn fromShippy, Appel and Shoemaker, 1984.)

BASIC PRINCIPLES 301
the extracellular space. In normal individualsapproximately 25% of the infused volume remainswithin the blood vessels when equilibrium is reached(usually within 20–30 minutes). In other words, forevery liter of infused crystalloid approximately750 ml will pass into the interstitium and 250 ml willremain in the plasma. Thus interstitial edema is anecessary consequence of volume resuscitation withcrystalloids and should not, unless excessive, beinterpreted as evidence of fluid overload. However,the volume of crystalloid used in resuscitatingpatients with head injury should be tempered by thepossibility of worsening brain edema.
Crystalloids are well suited for replacing extrac-ellular fluid losses (dehydration); they are also used toreplace blood loss, based on the notion that acutehemorrhage (or hypovolemia) causes an interstitialfluid deficit that must also be replaced. Indeed,crystalloids have proven effective in resuscitatingpatients following acute hemorrhage and continue tobe widely used for this purpose (Moss and Gould,1988; Dodge and Glass, 1982; Tranbaugh and Lewis,1985; Shackford, 1987; Horton, Landreau and Tuggle,1985; Lowe et al., 1979; Virgilio et al., 1979).
Crystalloids are non-toxic and do not produceanaphylactoid reactions.
(c) Water-equivalent solutions
Solutions of 5% dextrose or 4% dextrose in 0.18%isotonic saline are essentially water rendered isotonicto prevent local red-cell lysis at the infusion site. Atmost infusion rates insufficient dextrose is present toalter blood glucose levels, so the fluid is distributedevenly throughout total body water. Thus for everyliter of solution administered, two-thirds will enter theintracellular space and one-third the extracellularspace. Three-quarters of the extracellular fluid will bein the interstitium and one-quarter in the plasma.Thus about 8% only of the infused volume remains inthe circulation. For this reason 5% dextrose solution isthe fluid of choice for patients with ischemic heartdisease or congestive cardiac failure, since it does notexpand the circulation and increase the cardiac work-load.
Dextrose and cerebral ischemia
The observation that carbohydrates promote ischemicdamage in the central nervous system is not new butseems forgotten (Voll and Auer, 1988). The centralnervous system relies on glucose for much of itsenergy needs. When cerebral ischemia develops,glucose infusion will promote anaerobic glycolysisand produce large quantities of lactic acid, which,
accumulating locally, can reduce flow even further.Thus dextrose infusion during experimental cardio-pulmonary resuscitation was associated with a muchhigher mortality (Lundy et al., 1987). At this stage, theroutine infusion of glucose is not recommended inpatients at risk of cerebral insufficiency.
Other fluids such as 1 liter of half-isotonic 0.45%saline can be regarded as comprising 500 ml of isotonicsaline and 500 ml of free water.
(d) Hypertonic saline
Hypertonic saline will increase the osmolality of theECF, causing a water shift from cells to re-establishosmotic equilibrium. One liter of a 3% saline solutioncontains 510 mmol each of sodium and chloride – atotal of 1020 osmotically-active particles. These will beremain in the ECF, thus increasing its number ofosmotically active particles by 25% and its volume by7%. ECF osmolality will rise to 339 mmol/kg. To re-establish osmotic equilibrium, about 1.5 liters of waterwill shift from the ICF into the ECF. Thus the 1 liter ofa 3% saline solution will increase the ECF volume byabout 2.5 liters in total. As in other states of ECFexpansion, three-quarters of this volume (1.9 l) isdistributed to the interstitium and one-quarter (0.6 l)to the plasma, explaining the propensity of hypertonicsaline to cause pulmonary and peripheral edema(Table 16.4).
Hypertonic saline is effective in restoring volumeafter hemorrhage (Gala et al., 1991), endotoxic shock(Horton and Walker, 1991), trauma (Mattox et al., 1991;Vassar et al., 1991) and burn injury (Griswold et al.1991; Monafo, Halverson and Schechtman, 1984).Several clinical studies have suggested a better sur-vival after resuscitation with hypertonic solutionscompared with isotonic fluids (Mattox et al., 1991;Monafo, Halverson and Schechtman, 1984; Vassar etal., 1991).
Hypertonic solutions have a greater volume-expanding capacity than equal volumes of isotoniccrystalloid solutions. Approximately three times thevolume of 0.9% NaCl compared with 3.0% NaCl was
Table 16.4 Body compartment expansion according totype of resuscitation fluid: compartment expansion (ml)per 1000 ml of administered fluid
Fluid Plasma Interstitium Intracellular
Colloid 1000 0 0Isotonic (0.9%) saline 250 750 05% dextrose 83 250 667Half isotonic (0.45%) 167 500 133
saline3% saline 600 1900 –1500

302 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
required to restore MAP in a canine hemorrhagemodel, although the overall amount of sodium admin-istered did not differ between the two experimentalgroups. Because small infusion volumes cause rela-tively large increases in intravascular fluid volume,blood volume restitution is rapid; however this effectis short-lived (Gala et al., 1991).
Hypertonic solutions may reduce edema formationin non-injured tissues (Battistella and Wisner, 1991;Cross et al., 1988; Wisner, Schuster and Quinn, 1990).This may be particularly useful in head-injuredpatients. Several studies of resuscitation in experi-mental brain injury have found that hypertonicsolutions are more effective at lowering intracranialpressure and decreasing edema in the uninjuredcerebral hemisphere than isotonic fluids (Battistellaand Wisner, 1991; Wisner, Schuster and Quinn, 1990).In a study of trauma victims with concomitant headinjuries, hypertonic saline resuscitation was associatedwith increased survival compared with Hartmann’ssolution (Vassar et al., 1991).
At present hypertonic saline, with or without theaddition of colloid, is most commonly used forvolume resuscitation after trauma or burns. Theefficacy of small volumes and the rapidity of admin-istration make its use convenient. Other reportedbenefits of hypertonic compared to isotonic solutionsare improved pulmonary gas exchange (Boldt et al.,1991), increased renal cortical and gut mucosal bloodflow (Behrman et al., 1991), decreased bacterial trans-location (Reed et al., 1991a) and the induction ofnatriuresis (Gala et al., 1991) and kaliuresis (Battistellaand Wisner, 1991).
Although these properties would be advantageousin many clinical situations, the use of hypertonicsolutions is not without problems. In an experimentalmodel, increased bleeding, probably due to vasodila-tation, was reported if hypertonic fluid was admin-istered within 15 minutes of injury (Krausz et al.,1992). If more than 10% of normal plasma volume isreplaced with hypertonic saline, increases in pro-thrombin time and activated partial thromboplastintime and a decrease in platelet aggregation areobserved (Reed et al., 1991b). If large volumes ofhypertonic saline are used, potential complicationsinclude hypernatremia, hyperchloremia, hyperosmo-larity, hypokalemia, metabolic acidosis, intracellulardehydration, cerebral hemorrhage, inhibition of lipol-ysis, hyperosmolar coma and central pontine myeli-nolysis (Carvajal and Parks, 1988; Cross et al., 1988;Griffel and Kaufman,1992). These complications arerelated directly to the solute load infused andinversely to the patient’s volume of distribution.When using hypertonic saline serum sodium levels(less than 160 mmol/l) and serum osmolarity (lessthan 320 mosmol/l) require close monitoring.
16.4 Fluid resuscitation
16.4.1 SHOCK
When hypovolemia is severe, tissue oxygenationbecomes impaired and the clinical and metabolicfeatures of shock appear. The need for promptrestoration of adequate plasma volume under suchcircumstances is well recognized (Shoemaker, 1976;Shires and Canizaro, 1973). Hypotension has a mark-edly deleterious effect on the outcome from traumatichead injury and indeed from other injuries (Price andMurray, 1972; Siegel et al., 1991). Hence patients withhead injury should receive sufficient intravenous fluidresuscitation to avoid hypovolemia and hypotension(Buchman, Menker and Lipsett, 1991; Levison andTrunkey, 1982). Few issues in critical care medicinehave prompted as much diversity of opinion as themost appropriate asanguineous fluid to achieve thisgoal (Virgilio, Smith and Zarins, 1979; Shoemaker andHauser, 1979; Velanovich, 1989).
The primary abnormality in hypovolemic shock is areduction of plasma volume. Absolute or relativeplasma volume deficits contribute to the disturbancesof tissue oxygenation in the other three types ofcirculatory shock, cardiogenic, obstructive and septic,and these also may occur in the head-injured patient.Decreased plasma volume produces a decrease in leftventricular end-diastolic volume and stroke volume.Although sympathetic nervous system activation caninitially maintain cardiac output (CO) by inducingtachycardia and arterial pressure by producing vaso-constriction, at some point compensatory limits arereached and CO and systemic blood pressure maysuddenly fall.
There is clear agreement that the major goal oftreatment of circulatory shock associated with hypo-volemia is rapid restoration of blood volume andtissue oxygenation (Rackow et al., 1983; Weil andHenning, 1979). The use of asanguineous fluids forinitial resuscitation provides better restoration ofcapillary blood flow than does immediate transfusion.Moderate hemodilution, for example, to a hemoglobinlevel of 10–12 g/dl, is well tolerated by most patients,and does not lower DO2 if intravascular volume ismaintained (Messmer, 1975).
16.4.2 CHOICE OF FLUID
The hemodynamic response to fluid infusion is influ-enced by the choice of fluid, vascular tone and cardiaccompliance. Several studies have directly comparedthe plasma volume-expanding and hemodynamiceffects of colloids and crystalloids. In these studies, apreselected volume of fluid was administered ratherthan a volume adjusted on the basis of physiological
FLUID RESUSCITATION 303
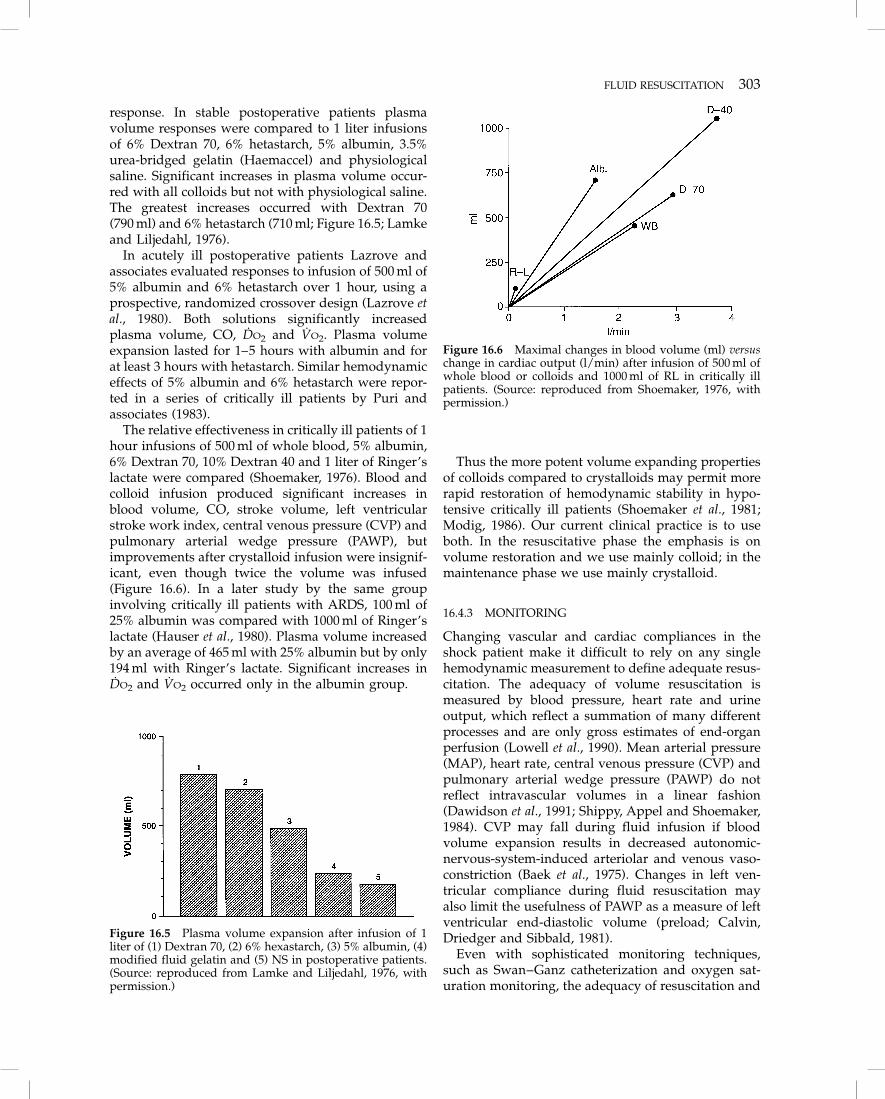
response. In stable postoperative patients plasmavolume responses were compared to 1 liter infusionsof 6% Dextran 70, 6% hetastarch, 5% albumin, 3.5%urea-bridged gelatin (Haemaccel) and physiologicalsaline. Significant increases in plasma volume occur-red with all colloids but not with physiological saline.The greatest increases occurred with Dextran 70(790 ml) and 6% hetastarch (710 ml; Figure 16.5; Lamkeand Liljedahl, 1976).
In acutely ill postoperative patients Lazrove andassociates evaluated responses to infusion of 500 ml of5% albumin and 6% hetastarch over 1 hour, using aprospective, randomized crossover design (Lazrove etal., 1980). Both solutions significantly increasedplasma volume, CO, DO2 and VO2. Plasma volumeexpansion lasted for 1–5 hours with albumin and forat least 3 hours with hetastarch. Similar hemodynamiceffects of 5% albumin and 6% hetastarch were repor-ted in a series of critically ill patients by Puri andassociates (1983).
The relative effectiveness in critically ill patients of 1hour infusions of 500 ml of whole blood, 5% albumin,6% Dextran 70, 10% Dextran 40 and 1 liter of Ringer’slactate were compared (Shoemaker, 1976). Blood andcolloid infusion produced significant increases inblood volume, CO, stroke volume, left ventricularstroke work index, central venous pressure (CVP) andpulmonary arterial wedge pressure (PAWP), butimprovements after crystalloid infusion were insignif-icant, even though twice the volume was infused(Figure 16.6). In a later study by the same groupinvolving critically ill patients with ARDS, 100 ml of25% albumin was compared with 1000 ml of Ringer’slactate (Hauser et al., 1980). Plasma volume increasedby an average of 465 ml with 25% albumin but by only194 ml with Ringer’s lactate. Significant increases inDO2 and VO2 occurred only in the albumin group.
Thus the more potent volume expanding propertiesof colloids compared to crystalloids may permit morerapid restoration of hemodynamic stability in hypo-tensive critically ill patients (Shoemaker et al., 1981;Modig, 1986). Our current clinical practice is to useboth. In the resuscitative phase the emphasis is onvolume restoration and we use mainly colloid; in themaintenance phase we use mainly crystalloid.
16.4.3 MONITORING
Changing vascular and cardiac compliances in theshock patient make it difficult to rely on any singlehemodynamic measurement to define adequate resus-citation. The adequacy of volume resuscitation ismeasured by blood pressure, heart rate and urineoutput, which reflect a summation of many differentprocesses and are only gross estimates of end-organperfusion (Lowell et al., 1990). Mean arterial pressure(MAP), heart rate, central venous pressure (CVP) andpulmonary arterial wedge pressure (PAWP) do notreflect intravascular volumes in a linear fashion(Dawidson et al., 1991; Shippy, Appel and Shoemaker,1984). CVP may fall during fluid infusion if bloodvolume expansion results in decreased autonomic-nervous-system-induced arteriolar and venous vaso-constriction (Baek et al., 1975). Changes in left ven-tricular compliance during fluid resuscitation mayalso limit the usefulness of PAWP as a measure of leftventricular end-diastolic volume (preload; Calvin,Driedger and Sibbald, 1981).
Even with sophisticated monitoring techniques,such as Swan–Ganz catheterization and oxygen sat-uration monitoring, the adequacy of resuscitation and
Figure 16.5 Plasma volume expansion after infusion of 1liter of (1) Dextran 70, (2) 6% hexastarch, (3) 5% albumin, (4)modified fluid gelatin and (5) NS in postoperative patients.(Source: reproduced from Lamke and Liljedahl, 1976, withpermission.)
Figure 16.6 Maximal changes in blood volume (ml) versuschange in cardiac output (l/min) after infusion of 500 ml ofwhole blood or colloids and 1000 ml of RL in critically illpatients. (Source: reproduced from Shoemaker, 1976, withpermission.)

304 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
oxygen delivery to individual tissue beds in vital endorgans cannot be determined. Poole et al. demon-strated this in an experimental canine shock model.Despite restoration of MAP and cardiac output (CO),cerebral blood flow did not return to preshock levels(Poole et al., 1986).
Nevertheless for clinical purposes the hemody-namic response to fluid resuscitation should beassessed by using the fluid challenge techniqueoriginally described by Weil and Henning (Table 16.5;Weil and Henning, 1979). When this is combined withsequential measurements of CO and measurements oftissue oxygenation (lactate, DO2, VO2 and mixedvenous oxygen saturation) the risks of under- andover-resuscitation are minimized (Shoemaker andCzer, 1979).
16.4.4 CRYSTALLOIDS VERSUS COLLOIDS
The basic disagreement that initiated the colloid versuscrystalloid controversy was a conceptual one. Thosefavoring crystalloids believe that an extracellular fluiddeficit plays a primary role in the pathophysiology ofhypovolemic shock and therefore repletion of thisdeficit is essential. Colloid proponents believe thatdecreased blood volume and reduced oxygen trans-port are the critical pathophysiological factors inhypovolemic shock; therefore, rapid and completerepletion of the intravascular compartment is criticalfor resuscitation.
In a series of experimental and clinical studiesinitiated in the early 1960s, Shires and associatesshowed that severe hemorrhagic shock was associatedwith a large decrease in extracellular volume, causedpredominantly by intracellular shift of interstitial fluidin addition to external losses or shift in the intra-vascular space (transcapillary refill; Shires et al., 1964,1972; Carrico, Canizaro and Shires, 1976). In a caninehemorrhagic shock model, animals resuscitated withshed whole blood alone or whole blood plus 10 ml/kgplasma had a significantly higher mortality than
animals resuscitated with whole blood and 50 ml/kgRinger’s lactate (Figure 16.7; Shires et al., 1964). In ababoon model of hemorrhagic shock, microelectrodeswere used to monitor transmembrane potential gra-dients across individual skeletal muscle cells. Sus-tained cell depolarization, a 49% decrease in extrac-ellular water, and 6% increase in intracellular waterincreased intracellular sodium and decreased intra-cellular potassium were all recorded, suggesting afailure of the ATPase-dependent Na+–K+ pump(Shires et al., 1972). Resuscitation with balanced saltsolution produced a return of the potential differenceto normal together with a decrease in intracellular andan increase in extracellular volume. Similar alterationsin water distribution were described in patients withcirculatory shock and those undergoing major oper-ative procedures (Shires, 1965; Shires, Williams andBrown, 1960). These changes also resolved withinfusion of balanced salt solutions.
Table 16.5 Guidelines for fluid challenge utilizing pulmonary arterial diastolic or pulmonary arterial wedge pressuremonitoring: 7/3 rule for fluid challenge. PAWP = pulmonary arterial wedge pressure (mmHg); PADP = pulmonaryarterial diastolic pressure (mmHg). (Source: adapted from Weil and Henning, 1979)
PAWP/PADP Fluid infusion rate
For 10 min before challenge < 12 200 ml � 10 min< 16 100 ml � 10 min≥ 16 50 ml � 10min
During 10 min infusion Change > 7 StopChange ≥ 3 Continue infusion without interruption
Immediately after 10 min infusion Change > 3, ≤ 7 Wait 10 min
After 10 min wait Change > 3 StopChange ≤ 3 Repeat fluid challenge
Figure 16.7 Survival after resuscitation of dogs fromhemorrhagic shock. Each group consisted of 10 dogs, bledinto shock by the Wiggers method. The RL group receivedfluid equivalent to 5% of body weight followed by return ofshed blood. The plasma group received 10 ml/kg of donorplasma plus shed blood. The blood group received shedblood alone. (Source: reproduced from Shires et al., 1964, ©1964, American Medical Association, with permission.)

EFFECTS OF INTRAVENOUS FLUIDS ON THE BRAIN 305
There is however, considerable controversy regard-ing the methods used by Shires and colleagues tomeasure extracellular fluid volume (Shoemaker, 1976;Roth, Lax and Malone, 1969). Using different tech-niques and models of hemorrhagic shock, other groupshave found either a minimal decrease in extracellularvolume accountable by the plasma volume loss or elsean increase in extracellular volume (Roth, Lax andMalone, 1969; Serkes and Lang, 1966; Moore et al.,1966). There is similar debate about the changes inextracellular volume after major surgery, where nosignificant change was found in patients after chol-ecystectomy and increases were found in patients aftercardiac surgery (Roth, Lax and Malone, 1969).
Despite this there is considerable evidence, fromexperimental models of severe hemorrhagic shock andfrom clinical studies of traumatic shock, that subjectscan be successfully resuscitated with balanced saltsolutions and blood (either whole blood or packed redblood cells) without the need for colloidal solutions(Lowe et al., 1979; Moss et al., 1981; Weaver et al., 1978).In the study of Lowe et al. (1979), patients withtraumatic abdominal injuries requiring laparotomywere resuscitated randomly with either Ringer’slactate or 4% albumin together with washed red bloodcells for the entire emergency room and intraoperativeperiod. Crystalloids alone were used postoperatively.Clinical criteria (systemic blood pressure, pulse andurine output) were used as the end points forresuscitation. Both groups of patients were resuscitatedsuccessfully and overall mortality was low (4.3%).There was no difference between the two groups in theneed for postoperative ventilatory support. Since mostpatients in this study were not in shock on admission,the results might not be applicable to patients who arein severe hemorrhagic shock. Moss et al. (1981) repeatedthe study in a group of patients with traumatic shock,defined as systolic arterial pressure of 80 mmHg or theneed for five or more red blood cell transfusions beforesurgery, with similar results. Pulmonary edema did notcomplicate the use of colloid or crystalloid in either ofthese studies. In both studies similar volumes of colloidand crystalloid were needed for resuscitation. Thissurprising finding may be due to the use of clinicalrather than physiological end points for fluid resuscita-tion. Whether the successful outcome with crystalloidresuscitation in these studies is related to replacementof an interstitial fluid deficit or to the limited amount ofthe infused solution that remains in the circulation hasnot been ascertained. Despite the numerous studiescomparing crystalloids to colloids, none has unequivo-cally demonstrated a distinct survival advantage witheither therapy.
The current recommendations of the AmericanCollege of Surgeons for initial resuscitation of patientswith traumatic injury (hemorrhagic shock) include
rapid infusion of up to 2 liters of Ringer’s lactate untilhemodynamic stability is restored. If the patientremains unstable packed red blood cells are theninfused. The use of two to four large-bore intravenouslines can compensate for the much more limitedvolume expansion produced by crystalloids. Compli-cations of this approach to fluid infusion are minimal(Moss et al., 1981).
The choice of fluids in patients with massivebleeding that is difficult to control, or those with pre-existing medical problems, is more controversial. Inthese situations fluid administration may result influid overload and subsequent morbidity. The risk ofpulmonary edema has been central to the debate overthe relative merits of crystalloids versus colloids. Asmentioned earlier, capillary membrane permeability,capillary hydrostatic pressure, colloid osmotic pres-sure (COP) and pulmonary lymph flow are allimportant factors in the development of pulmonaryedema. Despite claims to the contrary (Virgilio et al.,1979), weight gain and systemic edema due to fluidinfusion are not benign problems (Falk, 1991; Lowell etal., 1990). Decreased chest wall compliance due totissue edema may occur following hydration withcrystalloid (Brinkmeyer et al., 1981). Peripheral edema,particularly in a debilitated patient, can causedecreased mobility and subsequent skin breakdown.Tissue oxygenation is decreased when edema ispresent (Heughan, Niinikoski and Hunt, 1972) andwound healing may be impaired (Falk, 1991; Man-galore and Hunt, 1972; Niinikoski, Hengan and Hunt,1972).
Edema of the gastrointestinal tract may result inileus and intolerance for enteric alimentation (Falk,1991). The potential for bacterial translocation and thedevelopment of systemic sepsis and multiple systemsorgan failure may also be increased (Baker et al., 1988;Wilmore et al., 1988).
In addition, systemic edema can increase time onmechanical ventilation, require diuresis or dialysisand lengthen the stay in the ICU
16.5 Effects of intravenous fluids on thebrain
Parenteral fluid therapy, particularly in patients withhead injury, may increase brain swelling, intracranialpressure and neurological dysfunction and this maybe reflected in increased mortality and morbidity(Vassar et al., 1991; Battistella and Wisner, 1991; Falk,1991; Fein et al., 1982). However, there is littleinformation available on the relative effects of colloidor crystalloid administration on cerebral edema for-mation in normal subjects or in patients with braininjury and decreased intracranial compliance (Zornowet al., 1988).
306 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
16.5.1 THE EFFECTS OF CRYSTALLOIDS ANDCOLLOIDS ON THE NORMAL BRAIN
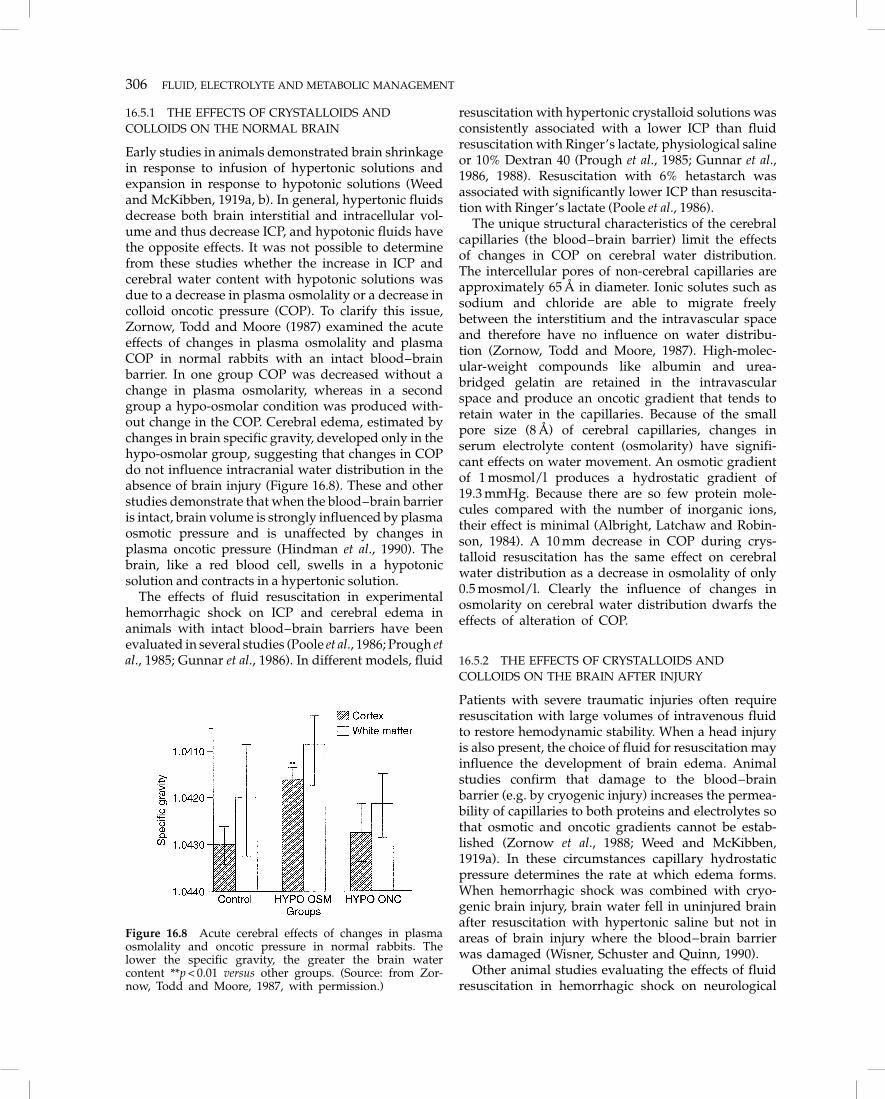
Early studies in animals demonstrated brain shrinkagein response to infusion of hypertonic solutions andexpansion in response to hypotonic solutions (Weedand McKibben, 1919a, b). In general, hypertonic fluidsdecrease both brain interstitial and intracellular vol-ume and thus decrease ICP, and hypotonic fluids havethe opposite effects. It was not possible to determinefrom these studies whether the increase in ICP andcerebral water content with hypotonic solutions wasdue to a decrease in plasma osmolality or a decrease incolloid oncotic pressure (COP). To clarify this issue,Zornow, Todd and Moore (1987) examined the acuteeffects of changes in plasma osmolality and plasmaCOP in normal rabbits with an intact blood–brainbarrier. In one group COP was decreased without achange in plasma osmolarity, whereas in a secondgroup a hypo-osmolar condition was produced with-out change in the COP. Cerebral edema, estimated bychanges in brain specific gravity, developed only in thehypo-osmolar group, suggesting that changes in COPdo not influence intracranial water distribution in theabsence of brain injury (Figure 16.8). These and otherstudies demonstrate that when the blood–brain barrieris intact, brain volume is strongly influenced by plasmaosmotic pressure and is unaffected by changes inplasma oncotic pressure (Hindman et al., 1990). Thebrain, like a red blood cell, swells in a hypotonicsolution and contracts in a hypertonic solution.
The effects of fluid resuscitation in experimentalhemorrhagic shock on ICP and cerebral edema inanimals with intact blood–brain barriers have beenevaluated in several studies (Poole et al., 1986; Prough etal., 1985; Gunnar et al., 1986). In different models, fluid
resuscitation with hypertonic crystalloid solutions wasconsistently associated with a lower ICP than fluidresuscitation with Ringer’s lactate, physiological salineor 10% Dextran 40 (Prough et al., 1985; Gunnar et al.,1986, 1988). Resuscitation with 6% hetastarch wasassociated with significantly lower ICP than resuscita-tion with Ringer’s lactate (Poole et al., 1986).
The unique structural characteristics of the cerebralcapillaries (the blood–brain barrier) limit the effectsof changes in COP on cerebral water distribution.The intercellular pores of non-cerebral capillaries areapproximately 65 Å in diameter. Ionic solutes such assodium and chloride are able to migrate freelybetween the interstitium and the intravascular spaceand therefore have no influence on water distribu-tion (Zornow, Todd and Moore, 1987). High-molec-ular-weight compounds like albumin and urea-bridged gelatin are retained in the intravascularspace and produce an oncotic gradient that tends toretain water in the capillaries. Because of the smallpore size (8 Å) of cerebral capillaries, changes inserum electrolyte content (osmolarity) have signifi-cant effects on water movement. An osmotic gradientof 1 mosmol/l produces a hydrostatic gradient of19.3 mmHg. Because there are so few protein mole-cules compared with the number of inorganic ions,their effect is minimal (Albright, Latchaw and Robin-son, 1984). A 10 mm decrease in COP during crys-talloid resuscitation has the same effect on cerebralwater distribution as a decrease in osmolality of only0.5 mosmol/l. Clearly the influence of changes inosmolarity on cerebral water distribution dwarfs theeffects of alteration of COP.
Figure 16.8 Acute cerebral effects of changes in plasmaosmolality and oncotic pressure in normal rabbits. Thelower the specific gravity, the greater the brain watercontent **p < 0.01 versus other groups. (Source: from Zor-now, Todd and Moore, 1987, with permission.)
16.5.2 THE EFFECTS OF CRYSTALLOIDS ANDCOLLOIDS ON THE BRAIN AFTER INJURY
Patients with severe traumatic injuries often requireresuscitation with large volumes of intravenous fluidto restore hemodynamic stability. When a head injuryis also present, the choice of fluid for resuscitation mayinfluence the development of brain edema. Animalstudies confirm that damage to the blood–brainbarrier (e.g. by cryogenic injury) increases the permea-bility of capillaries to both proteins and electrolytes sothat osmotic and oncotic gradients cannot be estab-lished (Zornow et al., 1988; Weed and McKibben,1919a). In these circumstances capillary hydrostaticpressure determines the rate at which edema forms.When hemorrhagic shock was combined with cryo-genic brain injury, brain water fell in uninjured brainafter resuscitation with hypertonic saline but not inareas of brain injury where the blood–brain barrierwas damaged (Wisner, Schuster and Quinn, 1990).
Other animal studies evaluating the effects of fluidresuscitation in hemorrhagic shock on neurological

METABOLIC RESPONSE TO INJURY 307
function have shown that, although ICP in animalsresuscitated with hypertonic saline was significantlylower, cerebral perfusion pressure was significantlyhigher in those receiving 6% hetastarch comparedwith those receiving hypertonic saline or physio-logical saline. Neurological function was also best inthe hetastarch group, suggesting that restoration ofcerebral perfusion pressure is a more important goalthan change in ICP alone. The vasodilatory effect ofhypertonic saline may have resulted in a lower meanarterial pressure compared with hetastarch.
These experimental results reaffirm that hypertonicsaline will shift water from the intracellular to theextracellular space. Ringer’s lactate is a hypo-osmolarsolution and 6% hetastarch is iso-osmolar; hence thediffering effects of these solutions on ICP can beexplained by their osmolarities.
The lower ICP after resuscitation with hypertoniccrystalloid solutions may assist in restoring of cerebralblood flow and DO2, but there is not universalagreement on this issue (Prough et al., 1986).
The days of keeping the patient with a head injury‘dry’ are over. There is no good evidence supportingfluid restriction as a means of limiting cerebral edemaafter brain injury (Morse et al., 1985). Dehydrationincreases sympathetic stimulation, metabolism and O2demand (Beckstead et al., 1978). Indeed, the ther-apeutic aim is now to maintain euvolemia and normalphysiological indices, especially cerebral perfusionpressure. Animal studies support this approach(Smith et al., 1982; Ito et al., 1979).
16.6 Metabolic response to injury
16.6.1 OVERVIEW
Major injury, whether surgical or accidental, evokespredictable metabolic, hormonal and hemodynamicresponses (Buckingham, 1985; Table 16.6). Changes incarbohydrate metabolism include increased endoge-nous hepatic glucose production (gluconeogenesis)and reduced glucose clearance (insulin resistance),which results in hyperglycemia. Fat becomes themajor body fuel; therefore lipolysis is increased andlipogenesis is retarded. Changes in protein metabo-lism are manifested by negative nitrogen balance
reflecting accelerated net protein breakdown (catabol-ism). The magnitude of these changes is proportionalto the extent of the injury (Weissman, 1990).
16.6.2 STARVATION
Starvation, i.e. lack of nutrient input, often accom-panies injury, while nutrient utilization is normal atthe cellular level. During starvation, metabolic adapta-tion occurs, with the aim of conserving essentialtissues. There is little or no activation of metabolicmediators and the system is still able to respond tonormal physiological stimuli. Glycogen reserves areonly small, approximately 200–400 g; thus liver glyco-genolysis is only useful for 24 hours. Decrease ininsulin and increase in glucagon secretion result inprotein and fat breakdown to provide energy. Aminoacids are converted to pyruvic acid, acetyl CoA andtricarboxylic acid (Krebs) cycle intermediates. Glyc-erol from fat is fed into the glycolytic pathway andfatty acids form acetyl CoA, some of which enters theKrebs cycle inside the mitochondria; some is con-verted to ketones, which are used by skeletal muscleand brain. Protein catabolism results in increasedurinary loss of urea, sodium, potassium, magnesiumand calcium. As the new energy pathways becomeestablished, protein breakdown decreases and fatbecomes the chief energy source, supplying 75–90% ofthe calories. The basal metabolic rate decreases as aresult of decreasing lean body mass, decreased phys-ical activity, and decreased thyroxine production.These changes are summarized in Figure 16.9.
In the absence of sepsis or injury, the starved patientcan usually be rapidly converted to the anabolic stateby administering moderate amounts of nutritionalsubstrate.
16.6.3 METABOLIC STRESS
When surgery, trauma or sepsis occur, a new process isactivated (Cerra, 1986). Injured or dead tissue, divid-ing organisms, severe perfusion deficits and resolvinghematomas activate mediator systems that affect end-organ function, producing the clinical, physiologicaland metabolic manifestations of the response to stress.This process is dynamic, and begins with a lag or ebbphase during which there is little metabolic activity.This is followed by a flow phase during whichmetabolic activity increases and peaks, usually on day3 or 4 after injury. The processes then abate overanother 3–4 days, unless a complication ensues. In theevent of a new stress, the process reactivates andreaches a new peak.
The resting energy expenditure rises to an amountthat depends on the type and severity of the stress.At high levels of stress, 30% or more of the increased
Table 16.6 Metabolic response to injury
� Altered protein homeostasis� Hypermetabolism� Altered carbohydrate metabolism� Sodium and water retention� Increased lipolysis

308 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
energy expenditure appears to come from the oxida-tion of amino acids, 30–40% from glucose and30–40% from fat. With activation of metabolic media-tors by trauma, gluconeogenesis becomes lessresponsive to exogenous nutritional substances (sec-tion 16.6.4(b)). Thus, administered glucose has pro-gressively less inhibiting effect on lipolysis, proteol-ysis and gluconeogenesis. Glucose calories, as well asother calories, which are in excess of the existingdemands promote lipogenesis, with excess CO2 pro-duction and hepatic dysfunction.
Proteolysis increases and amino-acid flux attemptsto meet the demands of energy production andprotein synthesis. Ureagenesis is increased and morenitrogen is present in the urine. Gluconeogenesis isincreased; peripheral glucose uptake is normal, butmuch of the glucose is recycled as lactate andalanine. Thus, increased plasma glucose and lactatelevels are a normal part of the response. Ketosis isrelatively depressed. These changes are summarizedin Figure 16.10.
Malnutrition usually develops rapidly in post-traumatic patients, taking days instead of the weeksneeded in simple starvation. It is difficult to excludethe effects of bedrest (disuse) on some of the para-meters. Indeed, in patients with isolated closed-headinjuries many reports have described persistentexcretion of large amounts of urinary nitrogen up toweeks after severe head injury. However some stud-ies indicate that the response abates in 5–7 daysunless a complication ensues. The early nitrogen
excretion appears to reflect the stress response,whereas the persistent nitrogen excretion may be dueto bedrest and disuse in a largely young and mus-cular group of patients.
In some instances under new stimuli the processreactivates and the state of persistent hypermetabol-ism with or without the development of multiplesystem organ failure ensues (Cerra, 1986). Risk fac-tors include severe perfusion shock, severe septicshock, persistent septic sources, and recurrent septicepisodes.
16.6.4 MEDIATORS OF THE RESPONSE TO INJURY
(a) The neuroendocrine axis
The mechanisms initiating, regulating and sustainingthis response are not all identified. It is known thatinjured patients have elevated levels of the anti-insulin hormones: cortisol, glucagon, and the cate-cholamines. Insulin levels are usually elevated, butnot sufficiently to prevent the commonly notedhyperglycemia. Growth hormone, aldosterone andADH are also elevated. These elevations may in partbe neurally mediated via the hypothalamus (Humeand Egdahl, 1959). Patients with severe head injurymay develop abnormalities of fluid and electrolytebalance because of damage to the neuroendocrineaxis (e.g. diabetes insipidus following pituitary dam-age; Figure 16.11).
Figure 16.9 Acute non-stressed fasting metabolism. The initial flow of substrate is designed to provide glucose for obligateand non-obligate glucose users. With time, ketones and fats become the main energy substrates, resulting in daily urinarynitrogen excretion.

METABOLIC RESPONSE TO INJURY 309
(b) Cytokines
Several non-endocrine factors play important roles inthe response to stress. These include interleukin-1 (IL-1), tumor necrosis factor (TNF), IL-2 and gamma-interferon (IFN). The reader is referred to the extensivereview of this subject by Weissmann (1990).
16.6.5 EFFECT OF STRESS ON FLUID ANDELECTROLYTE MOVEMENTS
The changes in electrolyte concentrations and fluiddistribution after trauma and critical illness have beenextensively studied. In the normal subject neural,hormonal, hemodynamic and renal mechanisms func-tion in a highly integrated manner to preserve sodiumand water homeostasis. There are two main objectives.The first is to keep the concentration of sodium in theECF within a very narrow range. Together with itsassociated anions, sodium constitutes more than 90%of the total solute in the ECF and controls thedistribution of water between the cells and theextracellular space. Large deviations from normal inECF sodium concentration cause cells to shrink orswell, which may have serious consequences for brainfunction. Sodium concentration is kept constant byfinely adjusting the water content of the ECF throughthe secretion of antidiuretic hormone (ADH, vasopres-sin; Figure 16.12). At least four independent physio-logical stimuli release ADH into the blood stream:osmotic, hemodynamic (hypotension and/or hypovo-
lemia), nausea and emesis, and hypoglycemia. Ofthese, osmotic stimulation is the most important forfine control of ADH secretion and maintenance ofwater balance. A number of drugs used in critically illpatients affect ADH secretion. Halothane and mor-phine in high doses increase ADH release, whilephenytoin and chlorpromazine inhibit ADH release.Positive pressure ventilation also increases secretion.The thirst mechanism assists by controlling fluidintake to some extent in the conscious patient.
The second objective is to keep the total sodiumcontent of the ECF within normal limits and thusmaintain a normal ECF volume. Since sodium is themajor cation of the ECF and the body adjusts the wateraround it to maintain a normal sodium concentration,the total number of sodium ions in the ECF willdetermine the ECF volume. Large deviations fromnormal in the ECF sodium content cause fluctuationsin the circulating blood volume. Both volume contrac-tion and expansion can have serious effects on brainfunction, particularly in the presence of pre-existingbrain damage (Aubry and Nankin, 1965). Normally,ECF sodium is regulated by several closely coordi-nated mechanisms that adjust the amount of sodiumlost through the kidneys.
� The relative fullness of the ECF is sensed byreceptors located in low- (intrathoracic) and high-pressure (intra-arterial) areas of the cardiovascularsystem. When changes in ECF volume occur, renal,neural and hormonal mechanisms modulate renal
Figure 16.10 Summary of moderate to severe stress metabolism. The neuroendocrine and intrinsic mediator systemsmodulate the metabolic machinery to mobilize fat and amino acid stores, which provide a continuous supply of fuel to meetincreased energy demands and provide an increased supply of substrate for the production of stress proteins. Increasedurinary nitrogen excretion is one result of the process. BCAA = branched-chain amino acids.
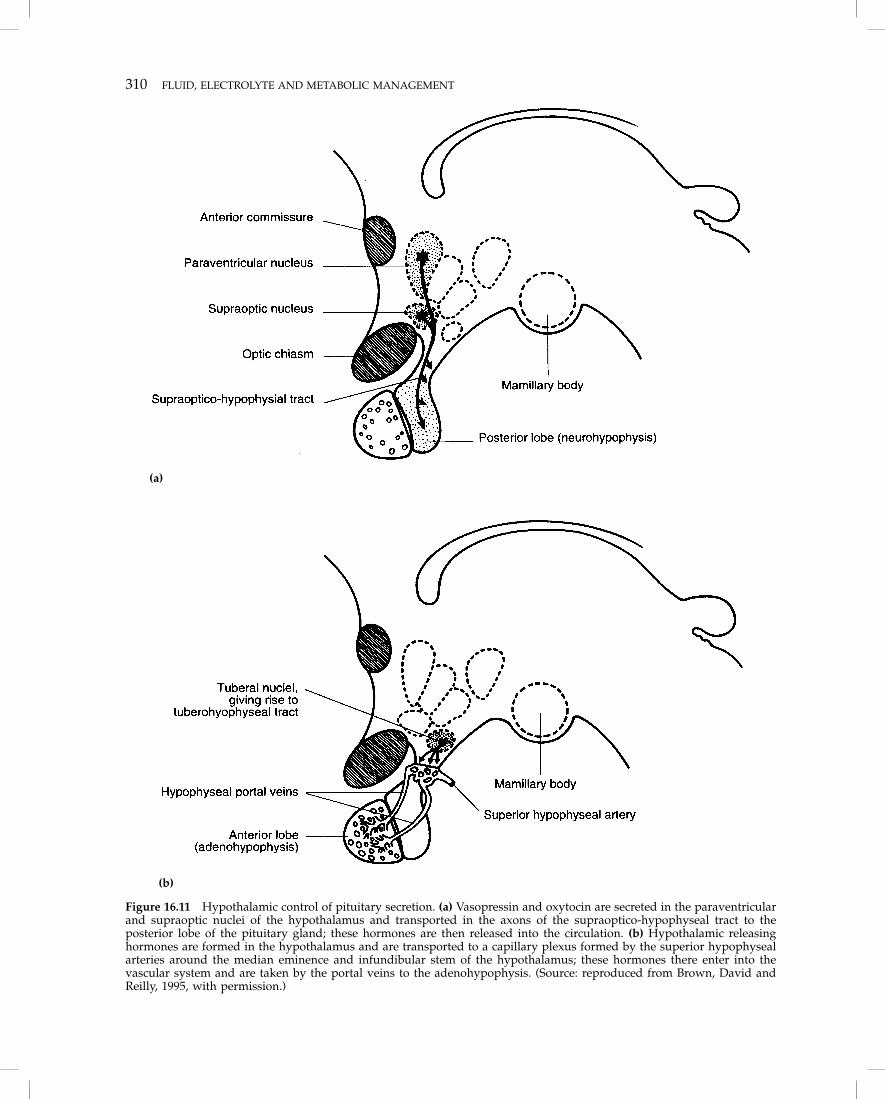
310 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
(a)
(b)
Figure 16.11 Hypothalamic control of pituitary secretion. (a) Vasopressin and oxytocin are secreted in the paraventricularand supraoptic nuclei of the hypothalamus and transported in the axons of the supraoptico-hypophyseal tract to theposterior lobe of the pituitary gland; these hormones are then released into the circulation. (b) Hypothalamic releasinghormones are formed in the hypothalamus and are transported to a capillary plexus formed by the superior hypophysealarteries around the median eminence and infundibular stem of the hypothalamus; these hormones there enter into thevascular system and are taken by the portal veins to the adenohypophysis. (Source: reproduced from Brown, David andReilly, 1995, with permission.)
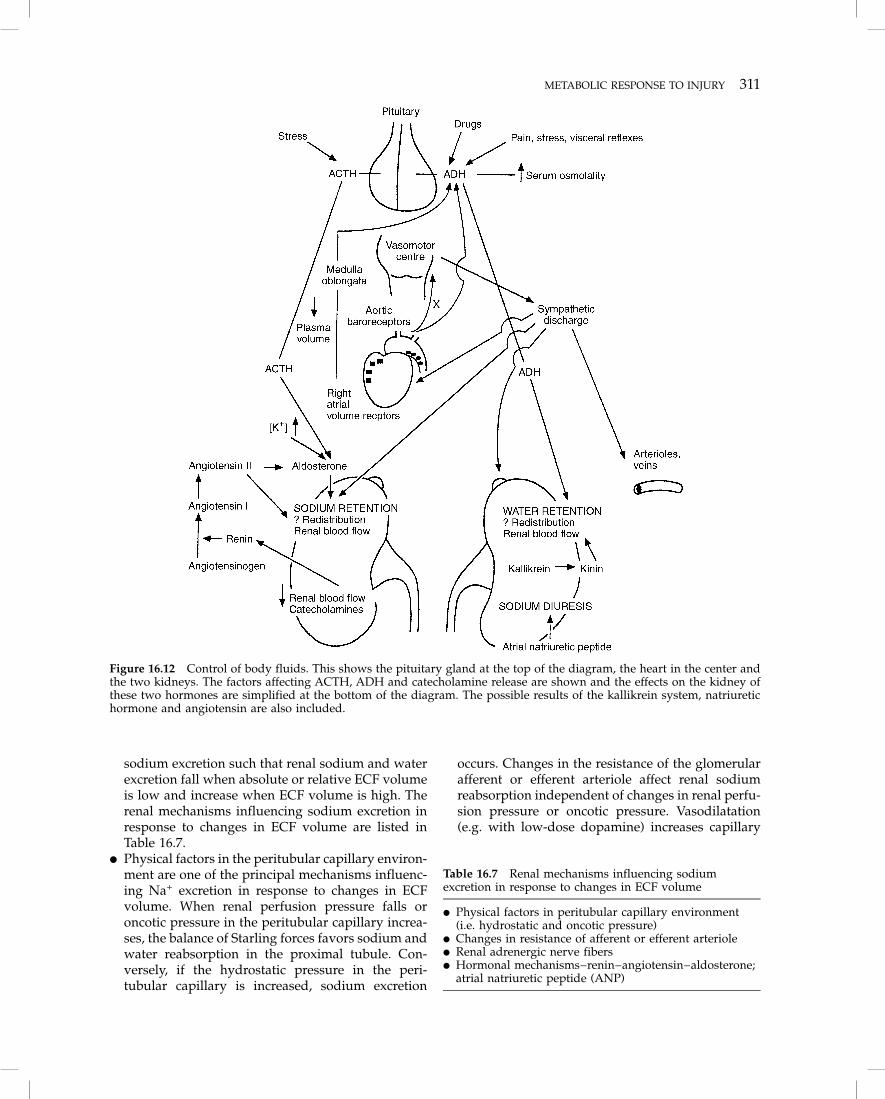
METABOLIC RESPONSE TO INJURY 311
sodium excretion such that renal sodium and waterexcretion fall when absolute or relative ECF volumeis low and increase when ECF volume is high. Therenal mechanisms influencing sodium excretion inresponse to changes in ECF volume are listed inTable 16.7.
� Physical factors in the peritubular capillary environ-ment are one of the principal mechanisms influenc-ing Na+ excretion in response to changes in ECFvolume. When renal perfusion pressure falls oroncotic pressure in the peritubular capillary increa-ses, the balance of Starling forces favors sodium andwater reabsorption in the proximal tubule. Con-versely, if the hydrostatic pressure in the peri-tubular capillary is increased, sodium excretion
occurs. Changes in the resistance of the glomerularafferent or efferent arteriole affect renal sodiumreabsorption independent of changes in renal perfu-sion pressure or oncotic pressure. Vasodilatation(e.g. with low-dose dopamine) increases capillary
Figure 16.12 Control of body fluids. This shows the pituitary gland at the top of the diagram, the heart in the center andthe two kidneys. The factors affecting ACTH, ADH and catecholamine release are shown and the effects on the kidney ofthese two hormones are simplified at the bottom of the diagram. The possible results of the kallikrein system, natriuretichormone and angiotensin are also included.
Table 16.7 Renal mechanisms influencing sodiumexcretion in response to changes in ECF volume
� Physical factors in peritubular capillary environment(i.e. hydrostatic and oncotic pressure)
� Changes in resistance of afferent or efferent arteriole� Renal adrenergic nerve fibers� Hormonal mechanisms–renin–angiotensin–aldosterone;
atrial natriuretic peptide (ANP)

312 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
hydrostatic pressure and renal sodium excretion,while vasoconstriction (e.g. heart failure) reduceshydrostatic pressure and renal sodium excretion.Adrenergic nerve fibers innervate the renal tubulesand may influence renal sodium excretion inde-pendent of changes in renal hemodynamics. Stim-ulation enhances sodium reabsorption, while dener-vation reduces renal Na+ reabsorption.
� The major hormonal mechanism influencing renalsodium excretion is the renin–angiotensin–aldos-terone system. The juxtaglomerular apparatussenses a reduction in effective blood volume, reninis released from the kidney, ultimately increasingangiotensin II and aldosterone levels. Aldosteroneincreases distal tubular sodium reabsorption andpotassium excretion. Angiotensin II also increasessystemic vascular resistance and proximal tubularsodium reabsorption by increasing resistance in theefferent arteriole.
ANP is produced both in atrial myocytes and withinthe CNS is also important in body sodium regulation(Needleman and Greenwald, 1986; Samson, 1987). Itsactions tend to oppose those of ADH and angiotensin,and so it appears to play a role in regulating fluid andelectrolyte balance. ANP may play a role in localcontrol of brain volume as well as electrolyte andwater content. ANP is released into the systemiccirculation from the atria, stimulated by increasedintravascular volume or by increased atrial pressureindependent of intravascular volume, and under salt-loading conditions as with infusion of hypertonicsaline. ANP decreases renal vascular resistance andincreases glomerular filtration rate. It induces natriur-esis and diuresis and inhibits the renin–angiotensin–aldosterone axis. ANP also reduces systemic vascularresistance and blood pressure.
Fluid balance disturbances are common amongpatients with head injury and are often multifactorial.Hyponatremia has often been attributed to a cerebralsalt-wasting state or to inappropriate secretion ofantidiuretic hormone (SIADH; as noted earlier, amoderate degree of ADH secretion is part of theresponse to stress). However, hyponatremia may infact often be iatrogenic (Bouzarth et al., 1982; Nelson etal., 1981). Hypernatremia and hyperosmolarity arepresent in many head-injured patients as a con-sequence of the fluid restriction and hyperosmolaragents used to manage the increases in intracranialpressure. Hypothalamic dysfunction can cause diabe-tes insipidus with hypernatremia.
The major fluid/electrolyte changes immediatelyafter acute injury are:
� release of K+ by the cells, leading to transientrelative hyperkalemia, increased K+ excretion andsubsequent reduction of total body K+;
� retention of Na+ and water; water is retainedrelatively more than Na+, leading to dilutionalhyponatremia.
These changes are maximum on the first and secondday after injury and persist for 4–7 days. They aremore pronounced after severe trauma (Verney, 1954).
16.7 Fluid therapy in uncomplicatedpostoperative and post-traumatic states
16.7.1 FLUID AND ELECTROLYTE REQUIREMENTS
Abnormal fluid losses occur through:
� external routes, such as hemorrhage, GI losses,wounds and burns; CSF otorrhea in young childrencan sometime be clinically significant;
� internal shifts into the interstitial water or extra-cellular water;
� local edema due to surgical wounds, crushinginjuries, burns, venous thrombosis;
� fluid accumulation in the transcellular space, e.g.pleural effusion, ascites and paralytic ileus.
Fluid replacement must take into account basal losses,additional extrarenal losses and distributional chan-ges, based on an understanding of the underlyingphysiological and metabolic changes.
Once a stable circulation has been established, waterreplacement in adult patients with no oral intakeshould equal urinary losses, usually 1000–1500 ml/d,plus insensible water losses, usually 600–800 ml/d,minus the volume of endogenous water released bytissue breakdown during semistarvation. In uncom-plicated, afebrile postoperative patients, the latter isusually only 100–200 ml but may be as much as1000 ml in a critically ill trauma patient with sepsis.Thus, replacement volume is approximately1500–3000 ml/d, depending on weight, age, sex andoverall status. Elderly patients with underlying car-diac or renal disease usually require less.
Water requirements increase by about 300 ml/d foreach 1°C elevation in body temperature. Fluid require-ments are greatly increased by shock, multipletrauma, sepsis, postoperative complications and othercritical illnesses. Under these conditions, criteria forfluid therapy are based primarily on hemodynamicfactors and adequate oxygen delivery.
Basal sodium requirements are met by the dailyadministration of 500 ml of physiological saline(75 mmol/d) plus the estimated volume of extrarenalfluid losses (e.g. nasogastric suction, diarrhea, CSFand other fistulae, and pleural or peritoneal drains).
In an uncomplicated postoperative patient, 40 mmolpotassium per day is usually sufficient, unless thepatient is suffering from renal failure or upper GIT

FLUID THERAPY IN UNCOMPLICATED STATES 313
bleeding. Such patients may develop hyperkalemiaand thus usually need less K+ replacement. Fluidtherapy of brief duration does not usually requiremagnesium, but debilitated patients who do not haverenal failure may benefit from 10–15 mmol Mg2+
daily.Nutritional support plays an integral role in the
management of metabolic stress. The time to initiatenutritional support is not completely settled. It isgenerally agreed that oxygen transport must berestored, stabilized and maintained prior to beginningnutritional therapy. In a previously well nourishedpatient, our practice is to wait a few days, whereas ina severely malnourished patient, therapy should beginas soon as O2 transport is stabilized.
16 7.2 NUTRITIONAL REQUIREMENTS
Nutrition may be administered via the enteral orparenteral route. The essential requirements are asfollows.
(a) Calories
Energy as carbohydrate or fat is given to meet themetabolic needs of the patient (i.e. basal metabolic rateplus additional demands). Sufficient energy of non-protein origin must be given to prevent infusedprotein being used as an energy source. Energy ismeasured in kilocalories (kcal) or kilojoules (kJ) – onekilocalorie = 4.184 kilojoules. A satisfactory ratio ofnon-nitrogen kilocalories per gram of nitrogen is 150:1(each 6.25 g protein yields 1 g nitrogen).
Although numerous equations and nomogramshave been used to estimate metabolic rate andnitrogen requirements, none predict accurately whichcaloric or nitrogen substrate will be utilized mosteffectively. Furthermore, substrate utilization variesduring the course of an illness.
The increase in metabolism during the acute phaseof injury is short-lived, and nutritional requirementsfall rapidly within 2–5 days as shock, pain and sepsisresolve and recovery begins. During convalescencevery few hospital patients expend more than1500–2000 kcal (6300–8400 kJ) per day.
Glucose
Glucose and lipid solutions are the standard intra-venous energy substrates. In a normal person a dailyinfusion of 4 mg/kg/min of glucose suppresses gluco-neogenesis maximally Approximately one-third of theinfused glucose is oxidized immediately and theremainder is stored as glycogen or lipid. Increasing theinfusion rate increases the amount of glucose stored,thus increasing O2 utilization and CO2 production. A
greater caloric intake is unnecessary in most patientsand may be detrimental in those with severe respira-tory failure, particularly if infusion rates of greater than7 mg/kg/min (equivalent to 700 g of dextrose or2800 kcal/d in a 70 kg subject) are used. Excessiveglucose administration may lead to hepatic steatosis,hyperbilirubinemia and an elevated alkalinephosphatase.
In patients requiring parenteral nutrition, glucoseshould be used as the major caloric source and infusedat a rate no greater than 30 kcal/kg/d (equivalent to500 g of dextrose or 2000 kcal/d in a 70 kg subject).Each gram of glucose provides approximately 4 kcal.
Lipid
Intravenous lipids are available as emulsions similarin particle size to chylomicrons, and are cleared assuch by the body. Chylomicrons are acted upon byplasma lipoprotein lipase to form free fatty acids andglycerol, which are either oxidized or stored as lipid.Although there is an increasing trend toward theroutine use of lipid solutions as an energy source,these expensive substances have no advantages overdextrose. Lipid solutions are only necessary to providethe essential fatty acids linolenic and linoleic. In theabsence of essential fatty acids, linoleic and hencearachidonic acid levels fall and skin lesions (desqua-matous dermatitis) may result. Approximately 9 kcalis provided for each gram of fat used by the body.
(b) Protein
The normal adult only needs an oral intake of 20 g ofprotein to meet daily protein requirements. Proteiningested in excess of this amount is broken down toprovide energy and nitrogenous wastes. In the acutelyill patient, the minimal quantity of protein required isunknown, but it is unlikely to exceed 50 g/d in apatient who is not losing protein externally. Theusually quoted requirement is 1–2 g/kg body weightper day.
Normal adults require 20 L-amino acids for proteinsynthesis, although only leucine, isoleucine, valine,lysine, threonine, phenylalanine, methionine and tryp-tophan cannot be synthesized and are thereforeessential. In patients requiring parenteral nutrition,histidine, arginine and cysteine may also berequired.
For effective nitrogen utilization, a ratio of essentialto total amino acids of 2:5 is advisable, and shouldreflect the amino acid profile of a normal diet. Toreduce the oxidation of amino acids as an energysubstrate, a source of alternate energy should beadministered simultaneously in a ratio of 150 kcal foreach 1 g of nitrogen.

314 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
(c) Vitamins and trace elements
Vitamins are dietary compounds that act as cofactorsfor intermediary metabolism. Patients may develop adeficiency of the water-soluble C and B group vitaminsand vitamin K within 4 weeks of parenteral nutrition;hence they should be administered to all patientsreceiving intravenous nutrition at doses equivalent tonormal daily requirement (Table 16.8). Vitamin defi-ciency may rarely affect neurological assessment.
The essential trace elements are those present in thebody in amounts of less than 50 �g/g tissue, and arerequired in the diet in milligram quantities or less perday. They are usually defined by their deficiency states,i.e. a reproducible functional and/or structural abnor-mality associated with a specific biochemical changewhich is prevented or reversed by the element.
With the exception of zinc, the body stores of theessential trace elements, copper, iodine, iron, man-ganese, cobalt, selenium, chromium and molybdenumare usually adequate for patients requiring parenteralnutrition for less than 3 months. A total of 2.5 mg of zincper day will normally maintain the zinc balance,although, in patients with diarrhea, up to five times thisamount may be required. Table 16.9 summarizesparenteral nutrient intake for adults. Trace elementdeficiency can result in neurological manifestations,which may interfere with the neurological assessmentof the head-injured patient and wound healing, as is thecase with some vitamin deficiency syndromes. There islittle data regarding the optimal amounts of provisionof trace elements to critically ill patients. Guidelineswill only be established when more accurate methodsare developed for assessing and monitoring traceelement status in patients in intensive care.
Gastric atony is usually present during states ofmetabolic stress and responds poorly to motilityenhancers. Gastric feeding is associated with a veryhigh incidence of aspiration and great care must betaken to avoid this. These problems are alleviated byusing sites for feeding distal to the pylorus and in ourview enteral nutrition should be used whenever
possible. In many patients the gut provides anadequate portal of entry for nutritional support at amuch reduced risk. By keeping bacteria and toxinswithin its lumen, the gut has a role in host defense. Instates of metabolic stress, especially coupled with gutstarvation, mucosal permeability increases, allowingmovement of enteric bacteria to the regional lymphnodes and thence to the systemic circulation.
Proven benefits of enteral feeding include main-tenance of an intestinal ‘barrier’ and better immunefunction, improved liver blood flow following shockand better wound healing. Gut feeding can be startedearly even in the presence of a mild ileus, althoughearly feeding does not attenuate the metabolicresponse to blunt trauma (Eyer et al., 1993). Exercise,even when done passively, is a potent anabolicstimulus and retards nitrogen mobilization in criti-cally ill and injured patients. While vigorous nutri-tional support appears to affect mortality favorablyand speed recovery from head injury, nutrient admin-istration alone cannot override the metabolic responseto head injury (Clifton et al., 1984; Young et al., 1985).Despite adequate caloric administration, severe head
Table 16.8 Recommended daily allowance (RDA) ofvitamins for adults (MVI-12 contents)
Vitamin RDA (mg) MVI-12(mg)
A 1 1D 0.005 0.005E 10 10K 0.1 –B1 (thiamin) 1.5 3B2 (riboflavin) 1.6 3.6B6 (pyridoxine) 2.2 4Niacin 18 40Folic acid 0.4 0.4B12 0.003 0.005Biotin 0.05 0.06Pantothenic acid 5 15Ascorbic acid 60 100
MVI-12 does not contain vitamin K
Table 16.9 Usual daily dose range of parenteral nutrient intake for the adult
Nutrient Amount Comment
Water 2500–3500 ml 30–40 ml/kg/dCalories 2500–3500 30–40 kcal/kg/dGlucose 600–900 gProtein equivalent (as amino acids) 60–130 g 1–2 g/kg/dNa+ 100–120 mmol ↑ with GI losses; ↓ in elderly, CCFK+ 80–120 mmol ↓ with renal failureCa2+ 4–10 mmolMg2+ 12–15 mmol ↑ with GI lossesPO4 10–15 mmol ↑ when glucose alone is given; ↓ with renal failure

DISORDERS OF WATER AND ELECTROLYTE BALANCE 315
(d) Nutrition and outcome
Since protein-calorie malnutrition can adversely affectthe structure and function of many organs, includingthe lungs (Arora and Rochester, 1982), heart (Heyms-field et al., 1978), gastrointestinal tract (James, 1971)and musculoskeletal system (Jeejeebhoy, 1986) as wellas wound healing (Haydock and Hill, 1987) andimmune function, nutritional support might be expec-ted to have a beneficial effect on mortality andmorbidity following head injury.
However, although there are studies that suggestthat nutritional support is beneficial in supportinggeneral metabolism following head injury (McMahonet al., 1993), a significant relationship between nutrientintake and outcome has not been proved.
16.8 Disorders of water and electrolytebalance
Hypovolemia (from aggressive use of diuretics),hyperosmolarity (from osmotic diuretics, hyperglyce-mia resulting in glycosuria, and diabetes insipidus)and disorders of sodium metabolism (especially thesyndrome of inappropriate ADH secretion – SIADH)are common problems in head-injured patients. Theseand other disorders may cause secondary brain injurythrough their effects on cerebral perfusion, intra-cranial pressure and neuronal function.
16.8.1 HYPONATREMIA
Hyponatremia (plasma Na+ < 135 mmol/l) is seen in5–12% of patients with severe head injury (Steinbokand Thompson, 1978). It is especially important,since the osmotic buffering capacity of the brain maybe impaired following primary brain injuries. ECFosmolality is reduced in all cases of hyponatremia inthe absence of abnormal solute accumulation, andeven small decreases in osmolality may cause brainedema, impairing brain function and increasing ICP.The pathogenesis of hyponatremia is summarized inTable 16.10.
Hyponatremia may occur in association with dif-ferent levels of body fluid tonicity (Table 16.11) andso be classified as isotonic, hypertonic or hypotonic,based on the measured plasma osmolality. Hypona-tremia following head injury is hypotonic and isusually thought to be due to SIADH. The risk ofhyponatremia seems greater in those with severehead injuries, chronic subdural hematoma and basalskull fracture (Steinbok and Thompson, 1978; Docziet al., 1982). There are numerous reports alleging that
this type of hyponatremia is due to renal salt wastingcaused by an unknown humoral substance releasedin response to cerebral injury (‘cerebral salt wasting’);however, the evidence is not strong and the reportedcases most probably represent SIADH (Oh and Car-roll, 1992). Deterioration in level of consciousness,new focal deficits, myoclonus, seizures or increasingICP should alert the physician to the possibility ofhyponatremia and hypo-osmolality in the patientwith a severe brain injury.
(a) Isotonic hyponatremia
An apparent decrease in serum sodium concentration(measured by flame photometry) occurs when themass of the non-aqueous components of serum isincreased by severe hyperlipidemia or hyperproteine-mia. This is usually referred to as pseudo-hypona-tremia and can be readily identified by finding anormal serum osmolality (Weisberg, 1989).
(b) Hypertonic hyponatremia
In patients with an increased amount of an imper-meant solute (i.e. one that is unable to cross the blood–
injury is followed by negative nitrogen balance,weight loss and depression of immune status and ofplasma protein levels.
Table 16.10 Pathogenesis of hyponatremia (ADH =antidiuretic hormone; GI = gastrointestinal)
� Shift of water from the cell (e.g. hyperglycemia,mannitol infusion)
� Shift of sodium into the cell (e.g. potassium loss)� Retention of water (e.g. syndrome of inappropriate
ADH secretion, edematous states)� Loss of sodium (e.g. through kidneys, GI tract, skin)
Table 16.11 Common causes of hyponatremia
Isotonic� Pseudohyponatremia
– Hyperlipidemia– Hyperproteinemia
Hypertonic� Hyperglycemia� Mannitol, glycerol or sorbitol excess
Hypotonic� Water retention
– Syndrome of inappropriate antidiuretic hormonesecretion
– Syndrome of inappropriate antidiuresis– Psychogenic polydipsia– Transurethral resection of the prostate (TURP)
syndrome� Salt depletion
– Adrenocortical failure– Diuretic excess
� Potassium depletion

316 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
brain barrier), such as glucose, mannitol, glycerol orsorbitol, osmotic equilibration occurs by water shift-ing from the ICF to the ECF, thus diluting the ECFsodium. In such circumstances, hyponatremia is oftenassociated with an elevated measured osmolality. Forevery 1 mmol/l rise in glucose above a plasma glucoselevel of 5.6 mmol/l, the plasma sodium decreases by0.288 mmol/l (Katz, 1973; Crandall, 1974; Jenkins andLarmore, 1974; Robin et al., 1979). The correctedsodium value is derived from the formula:
Corrected Na value = measured plasma Na+(plasma glucose value –5.6) � 0.288.
(c) Hypotonic hyponatremia
Hyponatremia is almost always caused by an excess oftotal body water. This may be due to excessiveinfusion of hypotonic or water-generating intravenousfluids such as 5% dextrose, 1.5% glycine or 0.45%saline, particularly when administered rapidly or inthe presence of high circulating ADH levels. Hypona-tremia may rarely be due to loss of exchangeablesodium or potassium (Fuisz, 1963).
Since hyponatremia may be associated with analteration in both total body water and total bodysolute the ECF volume may be increased (hyper-volemia), decreased (hypovolemia) or unchanged(isovolemia; Humes, 1986).
In health, a fluid intake of up to 15–20 liters may betolerated before water is retained. However, less wateris needed to produce hyponatremia in individuals inwhom there is also an increase in non-osmoticstimulation of ADH by hypovolemia, hypotension,pain, nausea or postoperative stress (Hayes, 1968;Chung et al.,1986; Sinnatamby et al., 1974), or areduction in urinary osmotic excretion, as in patientswith a reduced urea production, or a reduction inrenal capacity to respond to ADH.
The proposal that disease may cause a ‘sick cell’syndrome (Flear and Singh, 1973; Flear, Gill and Burn,1981) in which ECF sodium leaks into cells andproduces hyponatremia is a misconception (Leaf,1974; Bichet and Schrier, 1982).
Loss of potassium may cause hyponatremia assodium shifts into the cell in exchange for K+.
Syndrome of inappropriate ADH secretion (SIADH)
This syndrome is defined as hyponatremia due to anelevated level of ADH inappropriate for the prevailingosmotic or volume stimuli (Robinson, 1985; Bartterand Schwartz, 1967). ADH secretion from the neu-rohypophysis is no longer under normal regulatoryinfluences. The criteria for the diagnosis of SIADH arelisted in Table 16.12.
SIADH is a form of dilutional hyponatremia; ECFvolume is usually increased by 3–4 liters and periph-eral edema does not occur. Because of the expandedECF volume, glomerular filtration rate is increasedand the renin–angiotensin–aldosterone mechanism issuppressed, resulting in a decrease in the renalabsorption of sodium.
Syndrome of inappropriate antidiuresis (SIAD)
This syndrome is defined as hyponatremia due eitherto increased ADH secretion initiated by stimulation ofhigh- or low-pressure baroreceptors, or to a reductionin non-ADH renal water excretion mechanisms(Robertson, 1989). The causes of SIAD are listed inTable 16.13.
(d) Clinical features of hyponatremia
Hypotonic hyponatremia is almost always caused byexcess total body water, and the symptoms are due
Table 16.12 Criteria for the diagnosis of the syndrome ofinappropriate secretion of antidiuretic hormome (SIADH)
� Hypotonic hyponatremia� Urine osmolality greater than plasma osmolality� Urine sodium excretion greater than 20 mmol/l� Normal renal, hepatic, cardiac, pituitary, adrenal and
thyroid function� Absence of hypotension, hypovolemia, edema and
drugs� Correction by water restriction
Table 16.13 Causes of syndrome of inappropriateantidiuresis
Baroreceptor-mediated� Hypovolemia
– Addison’s disease– Hepatic failure– Nephrotic syndrome– Drugs: frusemide, thiazides
� Cardiac failure
Cortical influences� Pain, anxiety and nausea
– Postoperative– Trauma
Drugs� Narcotics, barbiturates, chlorpropamide, phenothiazines,
carbamazepine, tricyclics, nicotine derivatives, clofibrate,vincristine, vinblastine, cyclophosphamide
Reduction in renal response to antidiuretic hormone� Renal failure� Hypothyroidism� Addison’s disease� Drugs: indomethacin, frusemide, thiazides

DISORDERS OF WATER AND ELECTROLYTE BALANCE 317
to the cerebral water excess (cerebral edema; Waste-rlain and Posner, 1968). Normally the brain partiallyadapts to the hypo-osmolality within 24 hours,reducing the cerebral water excess by losing orinactivating intracellular osmotically active solutes(Arieff, 1984, 1985; Arieff and Guisado, 1976; Arieff,Liach and Massry, 1976).
If the patient develops hyponatremia of 125 mmol/l(osmolality 260 mosmol/kg) acutely, that is within 3days, then symptoms of cerebral edema usually occur.These include headache, anorexia, nausea, weakness,lethargy, confusion, disorientation, blurred vision,muscle cramps, coma and seizures (Arieff, 1984;Arieff, Liach and Massry, 1976; Plum and Posner,1980).
Chronic hyponatremia, on the other hand, withplasma sodium values well below 125 mmol/l, may bewell tolerated because of cerebral osmotic compensa-tion (Arieff, 1984). Thus the mortality associated withhyponatremia is usually related to its rate of develop-ment rather than its severity. Chronic hyponatremiahas a mortality of less than 10% (Sterns, 1987),whereas a mortality up to 50% has been reported withacute hyponatremia (Robertson, 1989).
(e) Diagnosis of hyponatremia
The evaluation of hyponatremia with hypo-osmolalitybegins with a clinical assessment of volume status andmeasurement of urinary indices (Table 16.14). How-ever, volume status can be difficult to assess clinicallyin a critically ill patient. ECF volume may be affectedby blood loss, the amount and type of fluid admin-istered and the use of diuretics. Urinary Na+ measure-ments are affected by the use of osmotic and non-osmotic diuretics. Figure 16.13 summarizes ourdiagnostic approach to hyponatremia.
Acute hyponatremia is usually due to excess water,as in dextrose solutions administered inappropriatelyto patients in the postoperative period. Diagnosis isbased on measurement of plasma osmolality, osmolargap and urinary sodium. The osmolar gap is the
difference between the measured osmolality and thecalculated osmolality (2 � Na+ + glucose + urea, whereplasma sodium, glucose and urea are measured inmillimoles per liter; Worthley, Guerin and Pain, 1987).When there is an excess of total body water, urinarysodium is usually greater than 40 mmol/l and plasmaurea is low. In the rare case of hyponatremia due tosalt depletion, urinary sodium is usually less than20 mmol/l and the plasma urea and uric acid are oftenhigh (Humes, 1986). In patients with head injurieshyponatremia will almost always be associated with aurinary osmolality of greater than 100 mosmol/kg.The presumptive diagnosis of SIADH can be con-firmed if simply restricting fluids results in a reduc-tion of urinary Na+ losses and correction of hypona-tremia. The clinical picture most often confused withSIADH is a patient with isotonic fluid loss, usuallydue to diuretics, who is treated with hypotonic fluidreplacement. In this situation, the body preservesvolume at the expense of tonicity – a form ofhypotonic dehydration. It should be stressed that theabsence of an increase in ECF volume excludes adiagnosis of SIADH.
Chronic hyponatremia accompanies other diseasesand it is important to differentiate SIADH fromsodium depletion due to prolonged vomiting, adrenalinsufficiency or hypopituitarism.
(f) Treatment of hyponatremia
Treatment aims to remove excess water and treat ofthe underlying cause.
Chronic hyponatremia (> 3 days’ duration)
If the patient is asymptomatic and hyponatremia hasbeen present for more than 3 days, fluid restrictionand removal of any precipitating factor is often all thatis required (Cluitmans and Meinders, 1990). Fluidshould be restricted to less than 500 ml/d. The rate ofcorrection should be no greater than 12 mmol/l/d(0.5 mmol/l/h) and is continued only until the plasmasodium is 130 mmol/l (Laureno and Karp, 1988;Sterns, Riggs and Schochet, 1986; Plum and Posner,1980; Cluitmans and Meinders, 1990; Norenberg andPapendick, 1984). If fluid deprivation is difficult tosustain in patients with SIADH or SIAD then patientswith hyponatremia and chronic CCF may benefit froman ACE inhibitor added to frusemide (furosamide).This will inhibit the stimulation of thirst and ADHrelease by angiotensin II (Dzau and Hollenberg, 1984;Packer, Medina and Yushak, 1984). However, in thesepatients a direct ADH inhibitor such as phenytoin maybe of greater value (Mulinari et al., 1990). This has beenused to reduce ADH release from the hypophysis in
Table 16.14 Extracellular volume (ECV) and urinarysodium (uNa+) in hyponatremia and hypo-osmolality
Diagnosis ECVuNa+
(mmol/l)
Dehydration Decreased < 10
Congestive heart failure, cirrhosis Increased < 10
Renal disease, adrenal insufficiency,diuretics
Decreased > 25
SIADH Normal orincreased
> 25

318 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
patients with SIADH due to CNS disorders includinghead injury (Martinez-Maldonado, 1980).
Complications of treatment of chronic hyponatremia
In chronic hyponatremia the brain is less able totolerate rapid correction and may develop centralpontine and extrapontine myelinolysis if hypertonicsaline is used. The lesions of central pontine myelinol-ysis (CPM) are caused by the destruction of myelinsheaths in the center of the basilar portion of the ponsand may extend from the midbrain to the lower pons.The clinical features range from coma, flaccid quad-riplegia, facial weakness and pseudobulbar palsy tominor behavioral changes without focal findings. The
onset may be from one to several days after thehyponatremia has been corrected and may requireMRI to confirm the diagnosis (Brunner et al., 1990).
While an association with CPM and correction ofhyponatremia has been reported, the relationshipbetween CPM and the rapidity of correction ofhyponatremia has not been firmly established inclinical practice. In 170 cases of CPM, hyponatremiaoccurred in only 28% of patients, most of whom werecorrected slowly (Ayus, Krothapalli and Arieff, 1985),an observation that has been confirmed by others(Tormey, 1990). Nevertheless, there is persuasiveexperimental evidence for the association betweenrapidity of correction of chronic hyponatremia andCPM (Norenberg and Papendick, 1984).
Figure 16.13 Diagnostic approach to hyponatremia. Arrows indicate the direction of change; single and double arrowsindicate the magnitude of change. Iso = isotonic; N = normal; V = variable. In hypertonic hyponatremia due tohyperglycemia, the corrected plasma Na+ = plasma Na+ + (plasma glucose – 10)/3 mmol/l.

DISORDERS OF WATER AND ELECTROLYTE BALANCE 319
Acute hyponatremia (< 3 days duration)
In this condition, the rate of reduction of plasmasodium is 0.5 mmol/l/h or greater and/or the hypo-natremia is of less than 3 days duration (Cluitmansand Meinders, 1990). It is often associated withinappropriate intravenous fluid administration andwith the postoperative period (Arieff, 1986). The totalwater excess in adults ranges from 5–10 liters.
The rate of correction of acute hyponatremia shouldbe no greater than 2 mmol/l/h of sodium until theplasma level has increased to 120 mmol/l or by amaximum of 20 mmol/l during the first 24 hours(Arieff and Guisado, 1976; Ayus, Krothapalli andArieff, 1987, 1988).
This is achieved initially by intravenous administra-tion of hypertonic saline given at 50–70 mmol/h. It isimportant to note that the purpose of using hypertonicsaline is not to correct a saline deficit, since there is infact not a total Na+ deficiency, but rather the hyper-tonic saline draws water into the intravascular com-partment and reduces brain edema. A spontaneous orfrusemide-induced diuresis (e.g. by 20–40 mg intra-venously) is then required to excrete the water excess.When the plasma sodium has increased to 120 mmol/l, precautions should be taken to prevent the plasmasodium rising to greater than 130 mmol/l over thenext 24 hours. If the hyponatremia presents withconvulsions, then urgent correction of the cerebraledema using 250 mmol of hypertonic sodium chlorideover 10 minutes (equivalent to 500 ml of 20% man-nitol), has been used. This will immediately elevatethe plasma sodium in adults by about 7 mmol/l (Table16 15; Worthley and Thomas, 1986).
Complications of treatment of acute hyponatremia
The complications reported with the use of hypertonicsaline include congestive cardiac failure, cerebralhemorrhage – intracerebral and subdural, presumeddue to tearing of bridging veins – and CPM. To reducethe incidence of congestive cardiac failure, CVP orPAWP should be monitored throughout saline admin-istration. Cerebral hemorrhage will only occur if thereis an inappropriate administration of hypertonicsaline in normonatremic states (Finberg, Luttrell andRedd, 1959).
16.8.2 HYPERNATREMIA
Hypernatremia is defined as a plasma sodium greaterthan 145 mmol/l. It is always associated with hyper-osmolality and may be caused by water depletion,excessive administration of sodium salts or both(Table 16.16).
In water-depleted states, the thirst mechanismnormally stimulates a conscious individual to drinkwater. Thus water loss only produces hypernatremia ifthe patient is unconscious or otherwise physicallyunable to drink. Excessive administration of sodiumsalts is a rare cause of hypernatremia and usually onlyoccurs through therapeutic misadventure.
Hypernatremia is the usual cause of hyperosmolarstates in neurosurgical patients, although marked
Table 16.15 Treatment of severe hyponatremia (ACE =angiotensin-converting enzyme; ADH = antidiuretichormone)
Acute hyponatremia� Fluid restrict to 500 ml/d or less� Hypertonic saline (50–70 mmol/h)� Diuresis of 160 ml/h or greater� Rate of correction
– First day: no greater than 20 mmol/l/d (2 mmol/l/h)until plasma sodium 120 mmol/l
– Second day: attempt to keep plasma sodium between130 and 135 mmol/l
� Seizures: 100–250 mmol hypertonic saline over 10 min
Chronic hyponatremia� Fluid restrict to 500 ml/d or less� Rate of correction:
– No greater than 12 mmol/l/d (0.5 mmol/l/h) untilthe plasma sodium is 130 mmol/l
� Drugs– Lithium, demeclocycline– Phenytoin– ACE inhibitor– ADH inhibitor
Table 16.16 Causes of hypernatremia
Water depletionExtrarenal loss� Exposure� GIT lossesRenal loss� Osmotic diuresis
– Urea, mannitol, glycosuria� Diabetes insipidus
– Central (neurogenic)Post-traumatic, postoperativeMetastatic tumors, craniopharyngioma, pinealoma,
cystsMeningitis, encephalitisFat embolismGranulomas (TB sarcoid)Idiopathic
– NephrogenicCongenitalHypercalcemiaHypokalemiaDrugs: lithium, amphotericin BRenal diseases: chronic pyelonephritis; medullary
sponge kidney; polycystic kidney; analgesicnephropathy; postobstructive uropathy; multiplemyeloma; amyloid; sarcoid
Salt gain� Hypertonic saline or sodium bicarbonate

320 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
hyperglycemia may contribute to hyperosmolality instates of insulin resistance, such as in sepsis, or as aresult of inadequate insulin administration with par-enteral nutrition.
(a) Clinical features
Hypernatremia is usually symptomatic if plasmasodium is 155–160 mmol/l or greater (equivalent to anosmolality of 330 mosmol/kg or greater). This equatesto a water depletion of 6 liters or greater in a 70 kgman (Arieff, 1984). Clinical features include pyrexia,restlessness, weakness, paralysis, irritability, drowsi-ness, lethargy, confusion, tremor, hyper-reflexia, seiz-ures and coma (Ross and Christie, 1969). The signs ofhypernatremia are due mainly to increased effectiveosmolality, which reduces brain cell volume (Oh andCarroll, 1992). As already noted, hypernatremia ismuch better tolerated if it develops slowly. Increase ofthe brain volume may be accomplished by intra-cellular shift of sodium, potassium and chloride, andthe accumulation of organic solutes, mainly taurineand other amino acids (Lohr, McReynolds and Gri-maldi, 1988). Normalizing brain volume in chronichypernatremia therefore requires an increase in thetotal solute content of the brain. A sudden lowering ofthe osmolality of the ECF may cause a shift of waterinto the brain cells with resultant cerebral edema.
(b) Diabetes insipidus
Diabetes insipidus (DI) is characterized by polyuria(3–15 l/d) and polydipsia due to the complete orpartial failure of ADH secretion (central or neurogenicDI), or decrease in the renal response to ADH(nephrogenic DI; Table 16.16). Both situations result inan inability to concentrate urine and to conserve
solute-free water in the face of appropriate stimuli.Head injury is a common cause of central DI. A veryearly onset is characteristic of major hypothalamicdamage and is associated with a high mortality (Seckl,Dunger and Lightman, 1987).
Head-injured patients with fractures involving thebase of the skull and sella turcica appear to be atincreased risk of DI (Crompton, 1971; Edwards andClark, 1986). Diabetes insipidus is common inpatients prior to brain death. Traumatic DI is thoughtto be caused by a shearing injury to the pituitarystalk. The time of onset is variable, but it usuallyappears after 5–10 days (Kern and Meislin, 1984). Inmost cases of post-traumatic DI the onset is charac-terized by polyuria, hypernatremia and plasmahyperosmolality, sometimes as early as 12–24 hoursafter injury. If the damage is limited to the pituitaryor lower pituitary stalk, the DI may only be tran-sient, and this is usually the case. High stalk lesionsor injury to the hypothalamus usually cause perma-nent DI. Since most patients in ICU are unable tocontrol their own water intake, hemodynamic insta-bility may occur with untreated DI.
Nephrogenic DI is often mild because the patientcan usually produce an isotonic urine; thus thediuresis is limited to 3–5 l/d.
(c) Diagnosis
Every patient with hypernatremia may be consideredto have an inadequate water intake, either absolute orrelative. Excessive sodium administration as the causeof hypernatremia is usually obvious from the history.Whether hypernatremia is due to insufficient waterintake or to excessive water loss can be determined bymeasurements of urine osmolality (Figure 16.14).Normal concentration of urine (urine osmolality
Figure 16.14 Differential diagnosis of hypernatremia. Posm = plasma osmolality; DI = diabetes insipidus.

DISORDERS OF WATER AND ELECTROLYTE BALANCE 321
> 700 mosmol/l) suggests that the main problem isinsufficient water intake, with or without excessiveextrarenal water loss. Urine osmolality between700 mosmol/l and the osmolality of plasma suggestspartial central DI, osmotic diuresis, diuretic therapy,acquired (partial) nephrogenic DI or renal failure. Ahigh (> 30 ml/kg/h) urine volume may be the first signof diabetes insipidus, but is also seen in osmoticdiuresis. A urine osmolality below that of plasmasuggests either complete central DI or nephrogenic DI.
Laboratory investigations show hypernatremia, per-sistently hypotonic urine and low urine osmolality,usually less than 200 mosmol/l. In milder cases urineosmolality may not be below that of plasma, and maybe 300–600 mosmol/l. Patients with central DI areunable to increase urinary osmolality during fluidrestriction, but remain sensitive to exogenous ADH.Patients with partial DI have urinary volumes muchless than those with complete DI. Other causes ofpolyuria which should be considered in the head-injured patient include osmotic diuretics (mannitol,iodinated contrast-medium), severe hyperglycemiaand fluid overload. In contrast to DI, a solute diuresisis usually accompanied by a higher urinary osmolality(between 250 and 320 mosmol/l; Shucart and Jackson,1976).
(d) Treatment of hypernatremia
Hypotension due to a reduction in intravascular fluidvolume should be treated by isotonic saline infusionsto replace the intravascular volume and improvetissue perfusion before hypotonic solutions are used.
Pure water depletion is treated by water administra-tion. (In a conscious patient with normal gastro-intestinal function, oral ingestion of water is encour-aged.) If intravenous fluid is required, 5% dextrose orhypotonic saline solutions (e.g. 0.45% saline) are oftenused, as sterile water through a peripheral vein causeshemolysis (Krumbhaar, 1914). In rare cases, wheresaline and dextrose solutions are deemed inadvisable;for example, in a patient who is volume-depleted andseverely hypernatremic, central venous administra-tion of sterile water can be used without causinghemolysis (Worthley, 1986).
The rate of increase in osmolality should be nogreater than 2 mosmol/kg/h (Arieff, 1984; Arieff andGuisado, 1976). Hypernatremia due to excess sodiumis treated by water and diuretic administration(Addleman, Pollard and Grossman, 1985). The man-agement of DI depends on the cause (Table 16.17).
(e) Hyperosmolar therapy
Intravenous 20% mannitol (1098 mosmol/l) a six-carbon sugar similar to glucose, is often used to
lower ICP. The aim is not to induce hypovolemia,which may be detrimental, but rather to maintain anormovolemic state while increasing serum sodium,osmolality and tonicity, thus decreasing the extrac-ellular fluid volume within the brain. A serumsodium level of 145–155 mmol/l and serum osmolal-ity of 300–320 mosmol/l are often generated, andexcessive dehydration may precipitate prerenal renalfailure. Fluids are administered to maintain CVP
Table 16.17 Treatment of diabetes insipidus (DI) in thecritically ill ICU patient
Remarks
1 Confirm diagnosis Altered consious state or inabilityto detect thirst or take oral fluids.Hypertonicity (> 295 mosmol/kg),hypernatremia, inappropriatelydilute urine. DI may be transientor permanent; permanent formseen in one-third of patients withtrauma or neurosurgery. Presenceof associated anterior pituitaryhypofunction not uncommon inpatients with severe head trauma.
2 Replace free waterdeficit
Free H2O deficit (liters) =0.6 � (body weight [kg]) � ([Na+/140] – 1)Parenteral replacement of half ofdeficit immediately; rest over24–36 hours. Use 5% dextrose, notsaline solutions
3 Give maintenancefluids and replaceurine outputhourly withdextrose solutions
Labor-intensive; inaccurate fluidbalance may occur with highvolumes of urine
4 Daily weight Not always available in adult ICU
5 Close monitoringof electrolytes andfluid balance
6 Aqueousvasopressin (AVP)
If urine output remains excessive(> 200–250 ml/h) in the absence ofdiuretics or, if maintenance of fluidbalance is difficult orhyperosmolality is present give5–10 U subcutaneously or i.m.
7 Desmopressin(DDAVP) forpatients withsevere DI
More recent regimen. Syntheticanalog of AVP with longer half-lifeand fewer vasoconstrictive effects.Give 1–2 µg subcutaneously or i.v.every 12–24 h or by nasalinsufflation of 5–20 µg every 12 h.Watch for water intoxication(hyponatremia and elevatedurinary osmolality) duringtherapy; withhold DDAVPperiodically to documentpersistence of DI.

322 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
between 5 and 10 mmHg. In young, otherwise fithead-injured patients, pulmonary artery catheteriza-tion is rarely indicated.
Mannitol is administered as an intermittent intra-venous bolus (0.25–0.5 g/kg body weight) up to4-hourly. Where the blood–brain barrier is intact,mannitol remains within the intravascular and extrac-ellular spaces (Sutin, Ruskin and Kaufman, 1992). Ithas several effects, including an immediate increase incirculating blood volume and arterial blood pressure(Wise and Chater, 1962; Roberts et al., 1987). It alsoreduces blood viscosity by hemodilution (Burke et al. ,1981; Muizelaar et al., 1983) and by increasing redblood cell deformability. It causes osmotic dehydra-tion of the brain, which reduces the volume ofextracellular free water (Roberts et al., 1987), thusdecreasing brain volume and ICP as well as causing aurinary osmotic diuresis. Repeated administration ofmannitol may result in a systemic hyperosmolar stateand dehydration, which should be avoided sincecerebral perfusion may decrease and renal failure mayoccur. Repeated administration of mannitol may resultin its eventual movement through the blood–brainbarrier into the extracellular space, where theincreased oncotic pressure may retain free water andworsen edema. Thus mannitol use should be restrictedto the minimum amount needed to control ICP andCPP. Our practice is to measure serum osmolalitydaily and cease mannitol if serum osmolality exceeds320 mosmol/l.
Mannitol induces diuresis primarily by elevatingthe osmotic pressure of the glomerular filtrate to suchan extent that the tubular reabsorption of water andsolutes is hindered. It promotes the excretion ofsodium; however, proportionately more water thansodium is excreted. Excretion of potassium, chloride,calcium, phosphorus, magnesium, urea and uric acidare also increased. Mannitol is only very slightlymetabolized, if at all, to glycogen in the liver. It isfreely filtered by the glomeruli, with less than 10%tubular reabsorption, and is not secreted by tubularcells. The elimination half life in adults is about 100minutes. Approximately 80% of a 100 g dose isexcreted unchanged in the urine within 3 hours. In thepresence of renal disease in which glomerular functionis impaired or in conditions that impair small vesselcirculation, such as congestive cardiac failure, cir-rhosis with ascites, shock or dehydration, mannitolclearance is lower than normal. Mannitol does notcross the intact blood–brain barrier unless very highconcentrations are present in the plasma or the patienthas acidosis.
Hypertonic saline (7.5%) has also been used tolower ICP in animal models of head injury (Freshmanet al., 1993). It was as effective as 20% mannitol intreating elevated ICP caused by a space-occupying
lesion. Hypertonic saline has the additional benefit ofrapid small volume resuscitation of associated hemor-rhagic shock (section 16.5.2).
16.8.3 HYPOKALEMIA
Hypokalemia is defined as a serum potassium of lessthan 3.5 mmol/l or plasma potassium less than3.0 mmol/l. It may be due to a reduction in totalbody potassium, caused by a decreased oral intake,increased renal or gastrointestinal loss, or to a com-partmental shift of potassium from the ECF to theICF (Tannen, 1986). In contrast to Na+ and C1–,which are predominantly in the ECF and regulatedby the kidneys, K+ is predominantly in the ICF; itsmovements are primarily determined by nutritionaland metabolic factors (e.g. pH) and are mediated byneural and hormonal influences (Schultze and Nis-senson, 1980).
Hypokalemia may be caused by osmotic diuretics,diabetes insipidus, steroids, catabolic losses intrauma and secondary hyperaldosteronism (com-monly caused by hypovolemia or cardiac failure).Excessive renal K+ loss also occurs in the presence ofalkalemia and hypomagnesemia. Gastrointestinal K+
losses may be due to vomiting, nasogastric suction ordiarrhea. Intracellular shift of K+ occurs in metabolicor respiratory alkalosis, exogenous insulin admin-istration and endogenous or exogenous catechola-mine administration.
(a) Clinical features
The clinical features of hypokalemia are listed inTable 16.18. These include ventricular and supraven-tricular arrhythmias, ileus, enhancement of digitalistoxicity and hypokalemic nephropathy; nephropathyoccurs if hypokalemia is severe and prolonged andresults in impaired renal water conservation andpolyuria. If K+ depletion is severe neuromuscularmanifestations include weakness, hyporeflexia andeven paralysis. The muscular weakness can interferewith attempts to wean patients from ventilatorysupport in the ICU.
(b) Treatment
Treatment of hypokalemia is usually commencedwhen plasma K+ is less than 3.0 mmol/l. While there isnormally a linear relationship between serum K+ andtotal body K+ (Sterns et al., 1981), acid–base dis-turbances and other causes of compartmental K+ shiftmake estimates of total body K+ difficult in severely illpatients. Thus K+ therapy must be monitored byfrequent estimations of serum K+. Serum magnesiumlevels should be measured in patients with refractory

DISORDERS OF WATER AND ELECTROLYTE BALANCE 323
16.8.4 HYPERKALEMIA
Hyperkalemia is defined as a serum potassium greaterthan 5.0 mmol/l or plasma potassium greater than4.5 mmol/l. It may be due to excessive intake, severetissue damage, decreased excretion or body fluid
compartment shift, or may be artifactual, due tohemolysis after collection.
Hyperkalemia is uncommon in patients with anisolated head injury unless overzealous use of osmoticdiuretics has caused renal failure. Apparent hyper-kalemia may be seen with hemolysis or acidosis. Inpatients with additional trauma, rhabdomyolysis andmassive transfusion of old blood may result inhyperkalemia.
(a) Hypoaldosteronism (Barratt, 1981)
Hyporeninemic hypoaldosteronism
This usually occurs in patients who have non-insulin-dependent diabetes mellitus and mild renal failure,and may be provoked by cyclo-oxygenase inhibitors.The patient is hyperkalemic with metabolic acidosis(Type 4 renal tubular acidosis, RTA) and has anelevated plasma creatinine and urea nitrogen. Theetiology is not known, although hyporeninemia, pros-tacyclin deficiency and excess atrial natriuretic hor-mone have all been implicated. Non-steroidal anti-inflammatory drugs (NSAIDs), calcium-channelblockers and beta-adrenergic blockers should not beadministered as they will contribute to the hypor-eninemic state (Williams, 1986).
Primary hypoaldosteronism
This is characterized by normal or high renin levelsand is due to an enzymatic defect in the aldosteronepathway.
Pseudo-hypoaldosteronism
This is characterized by normal or high levels ofaldosterone with a renal tubular resistance to theaction of the hormone. This disorder is commonlycaused by spironolactone and amiloride. The suppres-sion of aldosterone production with hyperkalemia(Edes and Sunderrajan, 1985) described with heparinuse is due to the antiseptic in commercial heparinsolutions and not to heparin (Jaques, 1985).
(b) Clinical features of hyperkalemia
The clinical features of hyperkalemia include tingling,paresthesias, weakness, flaccid paralysis and hypoten-sion. ECG changes include T-wave peaking, P-waveflattening, PR prolongation (until sinus arrest withnodal rhythm occurs), QRS widening, shortened QTinterval and deep S wave. Finally, a sine-wave ECGpattern develops, which deteriorates to asystole,occurring at plasma potassium levels of 7 mmol/l orgreater.
Table 16.18 Clinical features of hypokalemia
Cardiac� Predisposition to digoxin toxicity� Ventricular and atrial tachycardias� Torsades de pointes� ECG changes: PR prolongation, T-wave inversion,
prominent U waves
Skeletal muscle� Weakness, hypotonicity� Ascending paralysis, ventilatory failure� Cramps, rhabdomyolysis
Gastointesinal� Constipation, ileus
Central nervous system� Depression, coma
Renal� Nephrogenic diabetes insipidus (polyuria, polydipsia)� Metabolic alkalosis� Interstitial nephritis� Predisposition to urinary tract infection
Endocrine� Glucose intolerance
hypokalemia, since both electrolytes may be disturbedtogether and hypokalemia may be difficult to correctwithout concomitant correction of hypomagnesemia(Tannen, 1986). Hypokalemia protects against hypo-calcemic tetany; thus hypocalcemia should be cor-rected before or together with the correction ofhypokalemia.
When hypokalemia is associated with metabolicalkalosis the standard treatment is intravenous or oralpotassium chloride. If the patient has renal tubularacidosis and hypokalemia, then potassium acetate isgiven. If phosphate depletion is also present, potas-sium phosphate may be administered. Phosphate canonly be infused slowly (at a maximum rate of2–4 mmol/h/70 kg ) and is usually given to correct aphosphate deficiency rather than potassium defi-ciency; thus potassium chloride is often givenconcomitantly.
Intravenous replacement of potassium should notnormally exceed 40 mmol/h. Plasma potassium levelsshould be monitored at 2–6-hourly intervals (Tannen,1986; Stockigt, 1977). Spironolactone, amiloride orACE inhibitors are more useful for preventing renalpotassium loss than for correcting an existing deficit(Tannen, 1986).

324 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
(c) Treatment
For mild hyperkalemia (i.e. plasma K+ > 4.5 and< 5.5 mmol/l) treatment usually involves reducing thepotassium intake, correcting renal failure, and treatingthe cause (e.g. ceasing NSAIDs or spironolactone).Treatment of severe hyperkalemia (i.e. plasmaK+ > 5.5 mmol/l) often involves the administration ofintravenous glucose and insulin, sodium bicarbonateand calcium chloride (Table 16.19; Tannen, 1986),although nebulized salbutamol (10–20 mg) may alsobe useful as it can reduce the plasma potassium by0.6–1.0 mmol/l after 30 minutes and will last for 2hours (Williams, 1986). Resonium A has the capacityto bind 3.1 mmol of potassium per gram by releasing3.1 mmol of sodium. Calcium chloride (or gluconate)does not reduce the plasma potassium and is usedonly to counteract the toxic cardiac effects of hyper-kalemia. Since all the methods of reducing thepotassium level are only temporary, treatment shouldalso be directed at the underlying cause, and may alsoinclude dialysis.
16.8.5 HYPOCALCEMIA
Hypocalcemia occurs in up to 70% of patients in ICU(Desai, Carlson and Geheb, 1988; Zaloga and Cher-now, 1986). It is defined as a total plasma calcium lessthan 2.2 mmol/l and may be caused by either areduced ionized or protein-bound calcium. In criti-cally ill patients calcium loss due to increased tissuesequestration of calcium may be associated withpancreatitis, septicemia, burns or toxic shock (Cher-now et al., 1982a; Zaloga, 1991). However, hypocalce-mia is only important clinically when the physio-logically active, ionized fraction (40% of total) isreduced. Ionized calcium is lowered by alkalosis, byhigh circulating free fatty acids, as in pancreatitis orsepsis, and by agents that chelate calcium, such asalbumin infusions, bicarbonate administration andhigh citrate loads administered with massive bloodtransfusion (Zaloga and Chernow, 1986). Ionizedhypocalcemia (less than 1.15 mmol/l) occurs in
15–20% of patients in the ICU (Zaloga, Chernow andCook, 1985) but cannot be predicted by estimation ofeither total serum calcium or of ionized calciumcorrected for pH and serum albumin concentration(Desai, Carlson and Geheb, 1987).
Common causes of hypocalcemia in head-injuredpatients include respiratory alkalosis, sepsis, hypo-magnesemia and rhabdomyolysis in the polytraumapatient. Hypomagnesemia impairs both parathyroidhormone (PTH) release and action. Critically illpatients who are septic or who have unexplainedionized hypocalcemia may be vitamin-D-deficient,have an impairment of vitamin D metabolism oraction, or have impaired release of PTH (Desai,Carlson and Geheb, 1987; Zaloga et al., 1984; Knochel,1977). Hyperphosphatemia in severe rhabdomyolysiscauses hypocalcemia by calcium precipitation orimpaired vitamin D metabolism.
(a) Clinical features
Clinical manifestations of ionized hypocalcemia aregenerally not evident until ionized calcium is less than0.7 mmol/l. Signs include hypotension, impaired ven-tricular function, bradycardia, bronchospasm andlaryngospasm. Weakness, tetany, hyper-reflexia, agi-tation, confusion and seizures may occur, but are lesscommon than in patients with hypoparathyroidism.The response to digoxin and catecholamines may alsobe impaired by hypocalcemia.
(b) Treatment
Treatment is only undertaken if the patient is symp-tomatic. Acute hypocalcemia may be corrected byintravenous 10% calcium chloride (0.7 mmol calciumper milliliter). Before initiating treatment, hypomag-nesemia should be excluded, as hypomagnesemicpatients respond poorly to calcium. Calcium chloride10%, 5 ml intravenously (i.e. 3.4 mmol) or 10 ml ofcalcium gluconate 10% (i.e. 2.3 mmol) may be admin-istered over 3–5 minutes. Rapid infusion of calciummay result in unpleasant flushing, hypertension,
Table 16.19 Treatment of life-threatening hyperkalemia
Agent Dose Onset (min) Duration (h)
IntravenousDextrose and insulin 50 g dextrose 10–15 3
20 IU insulinSodium bicarbonate 50–100 mmol 60–240 1–2Calcium chloride (10%) 5–10 ml (3.4–6.8 mmol) 1–5 1–2
Oral or rectal resonium A 50 g 120–240 3–12
Nebulized salbutamol 10–20 mg 30 2

DISORDERS OF WATER AND ELECTROLYTE BALANCE 325
bradycardia and atrioventricular nodal blockade, andmay precipitate digitalis toxicity. Persistent unex-plained hypocalcemia requires further evaluation toexclude hypoparathyroidism and vitamin Ddeficiency.
16.8.6 HYPOPHOSPHATEMIA
Hypophosphatemia is defined as a fasting plasmaphosphate level of less than 0.8 mmol/l. Moderatehypophosphatemia exists if the phosphate level isbetween 0.32 and 0.8 mmol/l (Bohannon, 1989).Severe hypophosphatemia exists when the plasmalevel is less than 0.32 mmol/l (Hayek and Eisenberg,1989). It may be caused by decreased intake, increasedexcretion or intracellular redistribution.
Parenteral or enteral nutrition induces a transcellularshift of phosphate from the ECF to the ICF via insulin-mediated intracellular transport of phosphate withglucose (‘refeeding syndrome’; Solomon and Kirby,1990; Aubier et al., 1985). Respiratory alkalosis causesintracellular shift of phosphate by stimulating glycol-ysis and intracellular phosphate trapping. Hypophos-phatemia induced by transcellular shift is associatedwith hypophosphaturia and the normal body stores ofphosphate are maintained. Hypokalemia and hypo-magnesemia may also promote hypophosphatemia byimpairing renal phosphate reabsorption.
(a) Clinical features
Clinical manifestations of hypophosphatemia are notusually seen unless levels are markedly reduced, i.e. toless than 0.3 mmol/l. Patients who are hyperventi-lated or malnourished, alcoholic, septic or havediabetic ketoacidosis are at increased risk of severehypophosphatemia, and diaphragmatic weakness andrespiratory failure may occur (Lau, 1986).
(b) Investigations
Patients with hypophosphatemia in the absence ofalkalosis and a urinary phosphate greater than25 mmol/d,are suffering excessive renal loss. If theurinary loss is less than 5 mmol/d, then renal loss isexcluded (Knochel, 1981). If hypophosphatemia isassociated with hypercalcemia, hyperparathyroidismshould be considered.
(c) Treatment
Hypophosphatemia is usually asymptomatic. If hypo-phosphatemia is due to compartmental shift resultingfrom respiratory alkalosis or to catecholamine infu-sions, even though phosphate levels may decrease to0.4 mmol/l, treatment with phosphate may not be
necessary. Generally, however, in the critically illpatient, the phosphate level should be kept above0.8 mmol/l, to ensure adequate respiratory, cardiac andintracellular function. Phosphate at 40–80 mmol/24 hmay be administered orally or intravenously as sodiumor potassium phosphate (Chernow et al., 1989). If anintravenous solution is administered as the dihydrogensalt, an equimolar amount of Na+ or K+ and 80% of themolar amount as H+ is administered with the phos-phate. If it is administered as the monohydrogen salt,then twice the molar amounts of sodium or potassiumare administered with the phosphate.
16.8.7 HYPOMAGNESEMIA
Hypomagnesemia is defined as a plasma level of lessthan 0.7 mmol/l and is associated with a 24-hour urinemagnesium of less than 1 mmol/l in the absence ofabnormal renal magnesium losses. This is a commonelectrolyte disturbance in ICU patients (Chernow et al.,1982b; Kingston, Al-Siba and Skooge, 1986; Paymaster,1976) and is caused by decreased intake or increasedloss. The causes are similar to those for hypophospha-temia. Common causes in patients with head injuryinclude diuretics (osmotic and non-osmotic), diabetesinsipidus, respiratory alkalosis, sepsis and certaindrugs including aminoglycosides.
(a) Clinical features
Clinical features tend only to occur when the plasmalevel is less than 0.5 mmol/l (Table 16.20; Paymaster,1976; Fishman, 1965; Whang et al., 1984). The clinicaldiagnosis of magnesium depletion is difficult becauseonly 1% of total body magnesium is contained inplasma and because total plasma magnesium does notaccurately reflect the ionized fraction (55% of thetotal), which is the physiologically active form. Theremaining plasma magnesium is bound to protein(33%) or is chelated. Measurement of ionized magne-sium is not available. Hypomagnesemia may beassociated with hypokalemia (due to renal loss),
Table 16.20 Clinical features of hypomagnesemia
Neurological� Confusion, irritability, delirium, convulsions� Ataxia, athetoid movements� Weakness, tremors, cramps, tetany
Cardiovascular� Tachyarrhythmias (torsades de pointes)� Enhanced digoxin toxicity
Biochemical� Resistant hypokalemia, hypocalcemia, renal tubular
acidosis
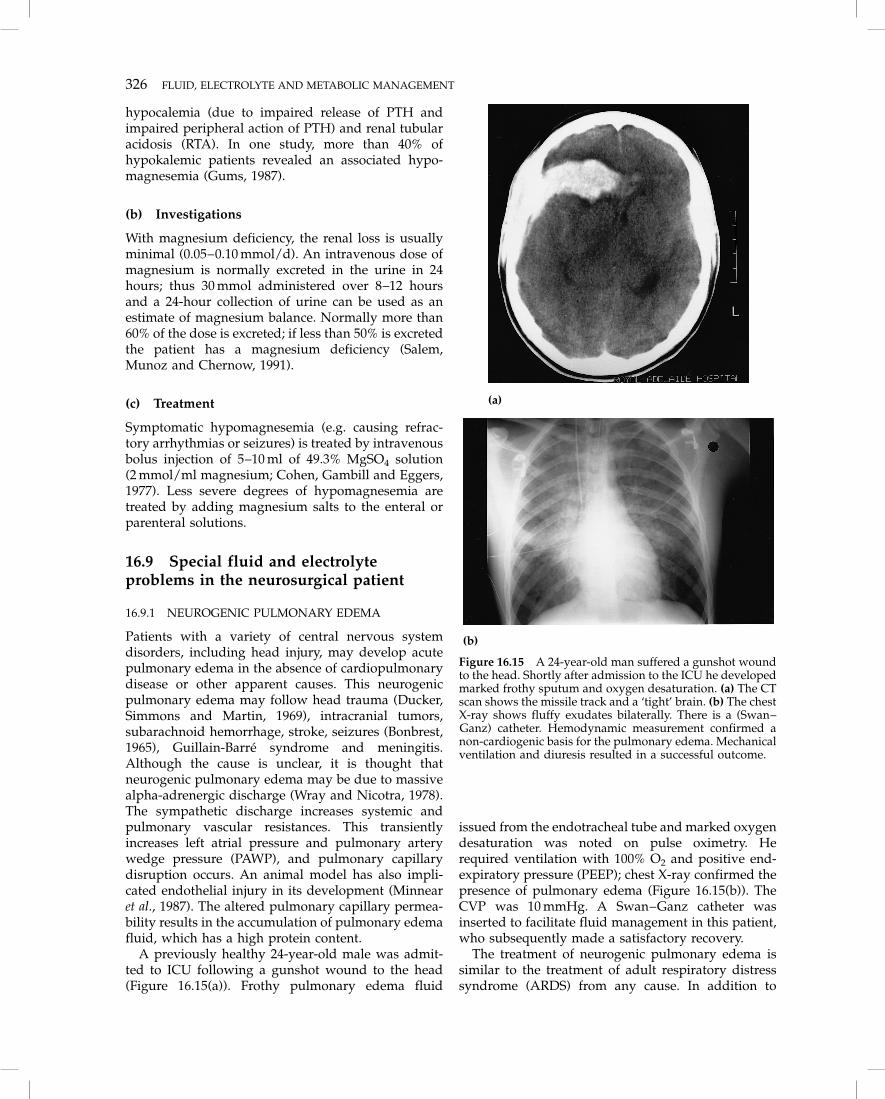
326 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
hypocalemia (due to impaired release of PTH andimpaired peripheral action of PTH) and renal tubularacidosis (RTA). In one study, more than 40% ofhypokalemic patients revealed an associated hypo-magnesemia (Gums, 1987).
(b) Investigations
With magnesium deficiency, the renal loss is usuallyminimal (0.05–0.10 mmol/d). An intravenous dose ofmagnesium is normally excreted in the urine in 24hours; thus 30 mmol administered over 8–12 hoursand a 24-hour collection of urine can be used as anestimate of magnesium balance. Normally more than60% of the dose is excreted; if less than 50% is excretedthe patient has a magnesium deficiency (Salem,Munoz and Chernow, 1991).
(c) Treatment
Symptomatic hypomagnesemia (e.g. causing refrac-tory arrhythmias or seizures) is treated by intravenousbolus injection of 5–10 ml of 49.3% MgSO4 solution(2 mmol/ml magnesium; Cohen, Gambill and Eggers,1977). Less severe degrees of hypomagnesemia aretreated by adding magnesium salts to the enteral orparenteral solutions.
16.9 Special fluid and electrolyteproblems in the neurosurgical patient
16.9.1 NEUROGENIC PULMONARY EDEMA
Patients with a variety of central nervous systemdisorders, including head injury, may develop acutepulmonary edema in the absence of cardiopulmonarydisease or other apparent causes. This neurogenicpulmonary edema may follow head trauma (Ducker,Simmons and Martin, 1969), intracranial tumors,subarachnoid hemorrhage, stroke, seizures (Bonbrest,1965), Guillain-Barre syndrome and meningitis.Although the cause is unclear, it is thought thatneurogenic pulmonary edema may be due to massivealpha-adrenergic discharge (Wray and Nicotra, 1978).The sympathetic discharge increases systemic andpulmonary vascular resistances. This transientlyincreases left atrial pressure and pulmonary arterywedge pressure (PAWP), and pulmonary capillarydisruption occurs. An animal model has also impli-cated endothelial injury in its development (Minnearet al., 1987). The altered pulmonary capillary permea-bility results in the accumulation of pulmonary edemafluid, which has a high protein content.
A previously healthy 24-year-old male was admit-ted to ICU following a gunshot wound to the head(Figure 16.15(a)). Frothy pulmonary edema fluid
issued from the endotracheal tube and marked oxygendesaturation was noted on pulse oximetry. Herequired ventilation with 100% O2 and positive end-expiratory pressure (PEEP); chest X-ray confirmed thepresence of pulmonary edema (Figure 16.15(b)). TheCVP was 10 mmHg. A Swan–Ganz catheter wasinserted to facilitate fluid management in this patient,who subsequently made a satisfactory recovery.
The treatment of neurogenic pulmonary edema issimilar to the treatment of adult respiratory distresssyndrome (ARDS) from any cause. In addition to
(a)
(b)
Figure 16.15 A 24-year-old man suffered a gunshot woundto the head. Shortly after admission to the ICU he developedmarked frothy sputum and oxygen desaturation. (a) The CTscan shows the missile track and a ‘tight’ brain. (b) The chestX-ray shows fluffy exudates bilaterally. There is a (Swan–Ganz) catheter. Hemodynamic measurement confirmed anon-cardiogenic basis for the pulmonary edema. Mechanicalventilation and diuresis resulted in a successful outcome.

REFERENCES 327
ventilatory support with positive-pressure ventilationand positive end-expiratory pressure (PEEP), fluidmanagement is of the utmost importance. Fluid intakeshould be crystalloid solution, rather than protein-containing solutions, and should be minimal. If PAWPis monitored, it should be kept low (5–7 mmHg) tominimize further extravasation of proteinacious fluid.If mean arterial pressure or perfusion is inadequate,inotropes should be used to maintain cerebral andperipheral tissue perfusion. For a detailed account ofthe respiratory management of neurogenic pulmonaryedema see Chapter 17.
16.9.2 NON-KETOTIC HYPERGLYCEMICHYPEROSMOLAR COMA
Non-ketotic hyperglycemic hyperosmolar coma(NHHC) may occur following head injury, intra-cerebral hemorrhage, brain tumor or cerebral infarc-tion (Park, Meacham and Netsky, 1976). About two-thirds of patients who develop NHHC have no historyof diabetes mellitus. Many have intercurrent infec-tions, or are taking drugs that alter glucose tolerance,such as steroids, phenytoin and thiazides, or aredehydrated.
The diagnosis of NHHC is based on the findings ofhyperglycemia, glycosuria and a plasma osmolalitygreater than 330 mosmol/l in the absence of ketosis.Patients who have NHHC and are dehydratedbecause of the osmotic diuresis are often potassium-depleted. They may also have evidence of prerenalrenal failure which, if untreated, can progress to acuterenal failure.
Treatment is primarily correction of dehydrationand hypertonicity. Physiological saline is used forvolume replacement, together with careful monitoringof serum and urine electrolytes and osmolality. Aftersodium deficits have been corrected with physio-logical saline, half physiological saline or dextrose/saline (4% dextrose in N/5 saline) should be used toreplace water deficits. The hyperglycemia is usuallyresponsive to insulin.
16.9.3 SPINAL-CORD-INJURED PATIENTS
Cervical cord injury may cause spinal shock due toanatomical or functional transection of the cord andthis may last for 1–3 weeks. The interruption tosympathetic outflow results in vasodilatation, poolingof blood in peripheral vascular beds and hypotension.Reflex bradycardia also occurs, because the cardiacaccelerator nerves arise from segments T1–T4. Thehypotensive phase is usually preceded by a rise insystemic pressure over 3–4 minutes as a result ofwidespread initial sympathetic activation. This maybe particularly important in the presence of brain
injury, because cerebral autoregulation may be over-come and cerebral edema exacerbated.
Because the heart cannot compensate for over-transfusion or increased venous return, careful mon-itoring is mandatory. Circulating volume is critical,and observation of the CVP or PAWP to fluidchallenge may be used to assess the volume status ofthe patient.
Electrolyte abnormalities may develop as a result ofimmobility and absent muscle activity. Mobilization ofcalcium may increase, causing hypercalcemia andhypercalciuria, from about 10 days after injury. Theincrease in serum calcium levels may predispose toventricular arrhythmias during anesthesia. Thus cal-cium levels should be measured preoperatively inthese patients.
16.10 Conclusions
� The goal of fluid therapy in the critically ill patientis to restore and maintain normal O2 delivery to thetissues before the sequelae of shock (includingsecondary neuronal damage) occur.
� Abnormalities of fluid, electrolyte and acid–basemetabolism may result in alterations in neurologicalstatus and thus interfere with neurological assess-ment, as well as contributing to secondary neuronaldamage and thus worsening outcome.
� Maintenance of normal cerebral perfusion pressure(CPP) is a key aspect of the management of patientswith head injuries, since impairment of cerebralautoregulation following traumatic brain injurycauses cerebral oxygen delivery to be influenced byCPP.
� Correction of hypovolemia is best achieved withcolloid, physiological saline or blood transfusion (ifnecessary). At least two animal studies have indi-cated that the amount of sodium infused does notadversely affect intracranial pressure (Ramming etal., 1994). In one of the studies, infusion of adextrose solution in amounts similar to the infusedsodium load significantly decreased serum sodium,increased brain edema and decreased neurologicaloutcome (Shapira et al., 1995).
16.11 ReferencesAddleman, M., Pollard, A. and Grossman, R. F. (1985) Survival after severe
hypernatremia due to salt ingestion by an adult. American Journal ofMedicine, 78, 176–178.
Adelson, E., Crosby, W. H. and Roeder, W. H. (1955) Further studies of ahemostatic defect caused by intravenous dextran. Journal of Laboratory andClinical Medicine, 45, 441.
Albright, A. L., Latchaw, R. E. and Robinson, A. G. (1984) Intracranial andsystemic effects of osmotic and oncotic therapy in experimental cerebraledema. Journal of Neurosurgery, 60, 481.
Alexander, B., Odake, K., Lawlor, D. et al. (1975) Coagulation, haemostasis andplasma expanders: a quarter century enigma. Federal Proceedings, 34, 1429.
Allen, S. J., Drake, R. E., Williams, J. P. et al. (1987) Recent advances inpulmonary oedema. Critical Care Medicine, 15, 963.

328 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
Arieff, A. I. (1984) Central nervous system manifestations of disorderedsodium metabolism. Clinics in Endocrinology and Metabolism, 13, 269–294.
Arieff, A. I. (1985) Effects of water, acid–base and electrolyte disorders on thecentral nervous system: hyponatremia, in Fluid, Electrolyte and Acid–baseDisorders, (eds A. I. Arieff and R. A. De Fronzo), Churchill Livingstone, NewYork, pp. 992–1007.
Arieff, A. I. (1986) Hyponatraemia, convulsions, respiratory arrest andpermanent brain damage after elective surgery in healthy women. NewEngland Journal of Medicine, 314, 1529–1535.
Arieff, A. I. and Guisado, R. (1976) Effects on the central nervous system ofhypernatremic and hyponatremic states. Kidney International, 10, 104–116.
Arieff, A. I., Liach, F. and Massry, S. G. (1976) Neurological manifestations andmorbidity of hyponatraemia: correlation with brain water and electrolytes.Medicine, 55, 121–129.
Arora, N. S. and Rochester, D. F. (1982) Respiratory muscle strength andmaximal voluntary ventilation in undernourished patients. AmericanReview of Respiratory Disease, 126, 5–8.
Atik, M. (1967) Dextran 40 and dextran 70. Archives of Surgery, 94, 664.Atik, M. (1969) The uses of dextran in surgery: a current evaluation. Surgery,
65, 548.Aubier, M., Murciano, D., Lecocguic, Y. et al. (1985) Effect of hypophosphate-
mia on diaphragmatic contractility in patients with acute respiratoryfailure. New England Journal of Medicine, 313, 420–424.
Aubry, R. H. and Nankin, H. R. (1965) Measurement of the osmotic thresholdfor vasopressin release in human subjects, and its modification by cortisol.Journal of Clinical Endocrinology and Metabolism, 25, 1481–1490.
Ayus, J. C., Krothapalli, R. K. and Arieff, A. I. (1985) Changing concepts intreatment of severe symptomatic hyponatremia. Rapid correction andpossible relation to central pontine myelinolysis. American Journal ofMedicine, 78, 897–902.
Ayus, J. C., Krothapalli, R. K. and Arieff, A. I. (1987) Treatment of symptomatichyponatremia and its relation to brain damage. New England Journal ofMedicine, 317, 1190–1195.
Ayus, J. C., Krothapalli, R. K. and Arieff, A. I. (1988) Correction ofhyponatraemia and its relation to brain damage. New England Journal ofMedicine, 318, 1336–1337.
Baek, S. M., Makabali, G. G., Bryan-Brown, C. W. et al. (1975) Plasmaexpansion in surgical patients with high central venous pressure (CVP): therelationship of blood volume to hematocrit, CVP, pulmonary wedgepressure and cardiorespiratory changes. Surgery, 78, 304.
Baker, J. W., Deitch, E. D., Ma, L. M. et al. (1988) Hemorrhagic shock inducesbacterial translocation from the gut. Journal of Trauma, 28, 896.
Barratt, L. J. (1981) Potassium handling by the kidney. Hyperkalaemia.Australian and New Zealand Journal of Medicine, 11, 16–22.
Bartter, F. C. and Schwartz, W. B. (1967) The syndrome of inappropriatesecretion of antidiuretic hormone. American Journal of Medicine, 42,790–806.
Battacharya, J., Gropper, M. A. and Staub, N. S. (1984) Interstitial fluidpressure gradient measured by micropuncture in excised dog lung. Journalof Applied Physiology, 56, 271.
Battistella, F. D. and Wisner, D. H. (1991) Combined haemorrhagic shock andhead injury: effects of hypertonic saline (7.5%) resuscitation. Journal ofTrauma, 31, 182.
Beckstead, J. E., Tweed, W. A., Lee, J. et al. (1978) Cerebral blood flow andmetabolism in man following cardiac arrest. Stroke, 9(6), 569.
Beecher, H. K. (1945) Preparation of battle casualties for surgery. Annals ofSurgery, 121, 769.
Behrman, S. W., Fabian, T. C., Kudsk, K. A. et al. (1991) Microcirculatory flowchanges after initial resuscitation of haemorrhagic shock with 7.5%hypertonic saline/6% dextran 70. Journal of Trauma, 31, 589.
Bichet, D. and Schrier, R. W. (1982) Evidence against concept of hypona-traemia and ‘sick cells’. Lancet, i, 742.
Bohannon, N. J. V. (1989) Large phosphate shifts with treatment forhyperglycaemia. Archives of Internal Medicine, 149, 1423–1425.
Boldt, J., Zickmann, B., Ballesteros, M. et al. (1991) Cardiorespiratoryresponses to hypertonic saline solution in cardiac operations. Annals ofThoracic Surgery, 51, 610.
Bonbrest, H. C. (1965) Pulmonary oedema following an epileptic seizure.American Review of Respiratory Disease, 91, 97–100.
Bouzarth, W. F., Shenkin, H. A. et al. (1982) Is cerebral hyponatraemiaiatrogenic? Lancet, i, 1061–1062.
Brinkmeyer, S., Safar, P., Motoyama, E. et al. (1981) Superiority of colloid overelectrolyte solution for fluid resuscitation (severe normovolemic hemodilu-tion). Critical Care Medicine, 9, 369.
Brobeck, J. R. (ed.) (1973) Best and Taylor’s Physiological Basis of Medical Practice,9th edn, Williams & Wilkins, Baltimore, MD, pp. 4–113–4-123; 1–15–1-29.
Brunner, J. E., Redmond, J. M., Haggar, A. M. et al. (1990) Central pontinemyelinolysis and pontine lesions after rapid correction of hyponatraemia: aprospective magnetic resonance imaging study. Annals of Neurology, 27,61–66.
Buchman, T. G., Menker, J. and Lipsett, P. A. (1991) Strategies for traumaresuscitation. Surgery, Gynecology, and Obstetrics, 172, 8–12.
Buckingham, J. C. (1985) Hypothalamo-pituitary responses to trauma. BritishMedical Bulletin, 41, 203–211.
Burke, A. M., Quest, D. O., Chein, S. and Cerri, C. (1981) The effects ofmannitol on blood viscosity. Journal of Neurosurgery, 55, 550–553.
Butterworth, J. F. and De Witt, D. S. (1989) Severe head trauma: pathophysiol-ogy and management, in Critical Care Clinics, vol. 5(4), (ed. D. S. Prough), W.B. Saunders, Philadelphia, PA, pp. 807–820.
Calvin, J. E., Driedger, A. A. and Sibbald, W. J. (1981) The hemodynamic effectof rapid fluid infusion in critically ill patients. Surgery, 90, 61.
Carrico, C. J., Canizaro, P. C. and Shires, G. T. (1976) Fluid resuscitationfollowing injury: Rationale for the use of balanced salt solutions. CriticalCare Medicine, 4, 46.
Carvajal, H. F. and Parks, D. K. (1988) Optimal composition of burnresuscitation fluids. Critical Care Medicine, 16, 695.
Cerra, F. B. (1986) The role of nutrition in the management of metabolic stress,in Critical Care Clinics, vol. 2(4), (ed. A. A. Meyer), W. B. Saunders,Philadelphia, PA, pp. 807–819.
Cervera, A. L. and Moss, G. (1975) Dilutional re-expansion with crystalloidafter massive haemorrhage: saline versus balanced electrolyte solution formaintenance of normal blood volume and arterial pH. Journal of Trauma, 15,498.
Chernow, B., Zaloga, G., McFadden, E. et al. (1982a). Hypocalemia in criticallyill patients. Critical Care Medicine, 10, 848–851.
Chernow, B., Smith, J., Rainey, T. G. et al. (1982b). Hypomagnesemia:Implications for the critical care specialist. Critical Care Medicine, 10,193–196.
Chernow, B., Bamberger, S., Stoiko, M. et al. (1989) Hypomagnesemia inpatients in postoperative intensive care. Chest, 95, 391–397.
Chung, H. M., Kluge, R., Schrier, R. W. and Anderson, R. J. (1986)Postoperative hyponatraemia. A prospective study. Archives of InternalMedicine, 146, 333–336.
Civetta, J. M. (1979) A new look at the Starling equation. Critical Care Medicine,7, 84.
Clifton, G. L., Robertson, C. S., Grossman, R. G. et al. (1984) The metabolicresponse to severe head injury. Journal of Neurosurgery, 60, 687–696.
Cluitmans, F. H. M. and Meinders, A. E. (1990) Management of severehyponatremia: Rapid or slow correction. American Journal of Medicine, 88,161–166.
Cohen, H. B., Gambill, A. F. and Eggers, G. W. N. (1977) Acute pulmonaryoedema following head injury: two case reports. Anesthesia and Analgesia,56, 136–139.
Consensus Development Conference (1988) Perioperative red blood celltransfusion. Report of consensus development conference on perioperativered cell transfusion. Journal of the American Medical Association, 260,2700–2703.
Crandall, E. D. (1974) Serum sodium response to hyperglycemia. New EnglandJournal of Medicine, 290, 465.
Crandall, E. D., Staub, N. C., Goldberg, H. S. et al. (1983) Recent developmentsin pulmonary oedema. Annals of Internal Medicine, 99, 808.
Crompton, M. R. (1971) Hypothalamic lesions following closed head injury.Brain, 94, 165–172.
Cross, J. S., Griber, D. P., Burchard, K. W. et al. (1988) Hypertonic saline fluidtherapy following surgery: a prospective study. Journal of Trauma, 29, 817.
Dawidson, I. J. A., Willms, C. D., Sandor, Z. F. et al. (1991) Ringer’s lactate withor without 3% dextran-60 as volume expanders during abdominal aorticsurgery. Critical Care Medicine, 19, 36.
Demling, R. H. (1980) Lung fluid and protein dynamics during haemorrhagicshock, resuscitation and recovery. Circulatory Shock, 7, 149.
Demling, R. H., Manohar, M., Will, J. A. et al. (1979) The effect of plasmaoncotic pressure on the pulmonary microcirculation after haemorrhagicshock. Surgery, 86, 323.
Demling, R. H., Harms, B., Kramer, G. et al. (1982) Acute versus sustainedhypoproteinaemia and post traumatic pulmonary oedema. Surgery, 92, 79.
Desai, T. K., Carlson, R. W. and Geheb, M. A. (1987) Parathyroid-vitamin Daxis in critically ill patients with unexplained hypocalcaemia. KidneyInternational, 32(22), S225.
Desai, T. K., Carlson, R. W. and Geheb, M. A. (1988) Prevalence and clinicalimplications of hypocalcaemia in acutely ill patients in a medical intensivecare setting. American Journal of Medicine, 84, 209–214.
Doczi, T., Tarjanyi, J., Huszka, E. and Kiss, J. (1982) Syndrome of inappropriatesecretion of antidiuretic hormone (SIADH) after head injury. Neurosurgery,10, 685–688.
Dodge, C. and Glass, D. D. (1982) Crystalloid and colloid therapy. Seminars inAnaesthesia, 1, 293–301.
Ducker, T. B., Simmons, R. L. and Martin, A. M. (1969) Pulmonary oedema asa complication of intracranial disease. American Journal of Diseases ofChildren, 118, 638–641.
Dzau, V. J. and Hollenberg, N. K. (1984) Renal response to captopril in severeheart failure: role of frusemide in natriuresis and reversal of hyponatremia.Annals of Internal Medicine, 100, 777–782.
Edelman, I. S. and Leibman, J. (1959) Anatomy of body water and electrolytes.American Journal of Medicine, 27, 256–263.
Edes, T. E. and Sunderrajan, E. V. (1985) Heparin-induced hyperkalemia.Archives of Internal Medicine, 145, 1070–1072.
Edwards, O. M. and Clark, J. D. A. (1986) Post-traumatic hypopituitarism.Medicine, 65, 281–290.

REFERENCES 329
Edwards, J. D., Nightingale, P., Wilkins, R. G. et al. (1989) Hemodynamic andoxygen transport response to modified fluid gelatin in critically ill patients.Critical Care Medicine, 17, 996.
Emerson, T. E. (1989) Unique features of albumin: a brief review. Critical CareMedicine, 17, 690.
Eyer, S. D., Micon, L. T., Konstantinides, F. N. et al. (1993) Early enteral feedingdoes not attenuate metabolic response after blunt trauma. Journal of Trauma,34(5), 639–644.
Falk, J. L. (1991) Fluid resuscitation and colloid-crystalloid controversy: newthoughts on an old debate. Critical Care Medicine, 19, 451.
Fein, I. A., Rackow, E. C., Sprung, C. L. et al. (1982) Relation of COP to arterialhypoxaemia and cerebral oedema during crystalloid volume loading ofpatients. Annals of Internal Medicine, 96, 570.
Finberg, L., Luttrell, C. N. and Redd, H. (1959) Pathogenesis of lesions in thenervous system in hypernatraemic states. Pediatrics, 23, 46–53.
Fishman, R. A. (1965) Neurological aspects of magnesium metabolism.Archives of Neurology, 12, 562–570.
Flear, C. T. G., Gill, G. V. and Burn, J. (1981) Hyponatraemia: mechanisms andmanagement. Lancet, ii, 26–31.
Flear, C. T. G. and Singh, C. M. (1973) Hyponatraemia and sick cells. BritishJournal of Anaesthesia, 45, 976–994.
Freshman, S. P., Battistella, F. D., Matteucci, M. et al. (1993) Hypertonic saline(7.5%) versus mannitol: a comparison for treatment of acute head injuries.Journal of Trauma, 35(3).
Fuisz, R. E. (1963) Hyponatremia. Medicine, 42, 149–168.Gala, G. J., Lilly, M. P., Thomas, S. E. et al. (1991) Interaction of sodium and
volume in fluid resuscitation after haemorrhage. Journal of Trauma, 31, 545.Gamble, J., Ross, G. and Tisdall, F. (1923) The metabolism of fixed base during
fasting. Journal of Biological Chemistry, 57, 633.Goldberg, H. S. (1980) Pulmonary interstitial compliance and microvascular
filtration coefficient. American Journal of Physiology, 239, H189.Granger, J. H. (1979) Role of the interstitial matrix and lymphatic pump in
regulation of transvascular fluid balance. Microvascular Research, 18, 207.Griffel, M. I. and Kaufman, B. S. (1992) Pharmacology of colloids and
crystalloids. Critical Care Clinics, 8, 235.Griswold, J. A., Anglin, B. L., Love, R. T. et al. (1991) Hypertonic saline
resuscitation: efficacy in a community-based burn unit. Southern MedicalJournal, 84, 692.
Gums, J. G. (1987) Clinical significance of magnesium: a review. DrugIntelligence and Clinical Pharmacology, 21, 240–246.
Gunnar, W., Merlotti, G. J., Jonasson, O. et al. (1986) Resuscitation fromhemorrhagic shock: Alterations of the intracranial pressure after normalsaline, 3% saline and dextran 40. Annals of Surgery, 204, 686.
Gunnar, W., Jonasson, O., Merlotti, G. et al. (1988) Head injury andhemorrhagic shock: Studies of the blood brain barrier and intracranialpressure after resuscitation with normal saline, 3% saline solution anddextran-40. Surgery, 103, 398–407.
Guyton, A. C., Granger, H. J. and Taylor, A. E. (1971) Interstitial fluid pressure.Physiology Review, 51, 527.
Harms, B. A., Kramer, G. C., Bodal, B. I. et al. (1981) Effect of hypoproteinae-mia on pulmonary and soft tissue oedema formation. Critical Care Medicine,9, 503.
Harrison, N., Darrow, D. and Yannet, H. (1936) The total electrolyte content ofanimals and its probable relation to distribution of body water. Journal ofBiological Chemistry, 113, 515.
Hauser, C. J., Shoemaker, W. C., Turpin, I. et al. (1980) Oxygen transportresponses to colloids and crystalloids in critically ill surgical patients.Surgery Gynecology and Obstetrics, 150, 811.
Haydock, D. A. and Hill, G. L. (1987) Improved wound healing response insurgical patients receiving intravenous nutrition. British Journal of Surgery,74, 320–323.
Hayek, M. E. and Eisenberg, P. G. (1989) Severe hypophosphataemiafollowing the institution of enteral feedings. Archives of Surgery, 124,1325–1328.
Hayes, M. A. (1968) Water and electrolyte therapy after operation. NewEngland Journal of Medicine, 278, 1054–1056.
Heughan, C., Niinikoski, J. and Hunt, T. K. (1972) Effect of excessive infusionof saline on tissue oxygen transport. Surgery, Gynecology and Obstetrics, 135,257.
Heymsfield, S. B., Bethel, R. A., Ansley, J. D. et al. (1978) Cardiac abnormalitiesin cachectic patients before and during nutritional repletion. American HeartJournal, 95, 584–594.
Hindman, B. J., Funatsu, N., Cheng, D. C. H. et al. (1990) Differential effect ofoncotic pressure on cerebral and extracerebral water content duringcardiopulmonary bypass in rabbits. Anesthesiology, 73, 951.
Holt, M. E., Ryall, M. E. and Campbell, A. K. (1984) Albumin inhibits humanpolymorphonuclear leucocyte luminol-dependent chemiluminescence: evi-dence for oxygen radical scavenging. British Journal of ExperimentalPathology, 65, 231.
Horton, J., Landreau, R. and Tuggle, T. (1985) Cardiac response to fluidresuscitation from haemorrhagic shock. Surgery, Gynecology, and Obstetrics,160, 444–452.
Horton, J. W. and Walker, P. B. (1991) Small volume hypertonic saline dextranresuscitation from canine endotoxin shock. Annals of Surgery, 214, 64.
Hume, D. M. and Egdahl, R. H. (1959) The importance of the brain in theendocrine response to injury. Annals of Surgery, 150, 697–712.
Humes, H. D. (1986) Disorders of water metabolism, in Fluids and Electrolytes,(eds J. P. Kokko and R. L. Tannen), W. B. Saunders, Philadelphia, PA, pp.118–149.
Ito, U., Ohno, K., Nakamura, R. et al. (1979) Brain edema during ischaemiaand after restoration of blood flow. Stroke, 10(5), 542.
James, W. P. T. (1971) Effects of protein-calorie malnutrition on intestinalabsorption. Annals of the New York Academy of Science, 176, 224–261.
Jaques, L. B. (1985) The new understanding of the drug heparin. Chest, 88,751–757.
Jeejeebhoy, K. N. (1986) Muscle function and nutrition. Gut, 27(Suppl.),25–39.
Jenkins, P. G. and Larmore, C. (1974) Hyperglycemia-induced hyponatremia.New England Journal of Medicine, 290, 573.
Jennett, B., Teasdale, G. and Braakman, R. (1979) Prognosis of patients withsevere head injury. Neurosurgery, 4, 283–289.
Karlson, K. E., Garzon, A. A., Shaftan, G. W. et al. (1967) Increased blood lossassociated with administration of certain plasma expanders: Dextran 75,dextran 40 and hydroxyethyl starch. Surgery, 62, 670.
Katz, M. A. (1973) Hyperglycemia-induced hyponatraemia – calculation ofexpected serum sodium depression. New England Journal of Medicine, 289,843–844.
Kee, D. B. and Wood, J. H. (1984) Rheology of the circulation. Neurosurgery,15(1), 125–131.
Kern, K. and Meislin, H. (1984) Diabetes insipidus: occurrence after minorhead trauma. Journal of Trauma, 24, 69–72.
Kingston, M. E., Al-Siba, M. B. and Skooge, W. C. (1986) Clinical manifesta-tions of hypomagnesemia. Critical Care Medicine, 14, 950–954.
Knochel, J. P. (1977) The pathophysiology and clinical characteristics of severehypophosphataemia. Archives of Internal Medicine, 137, 203–220.
Knochel, J. P. (1981) Hypophosphatemia. Western Journal of Medicine, 134,15–26.
Krausz, M. M., Landau, E. H., Klin, B. et al. (1992) Hypertonic saline treatmentof uncontrolled hemorrhagic shock at different periods from bleeding.Archives of Surgery, 127, 93.
Krumbhaar, E. B. (1914) Hemolysis due to intravenous injection of distilledwater. Journal of the American Medical Association, 62, 992–996.
Lamke, L. O. and Liljedahl, S. O. (1976) Plasma volume changes after infusionof various plasma expanders. Resuscitation, 5, 93–100.
Lau, K. (1986) Phosphate disorders, in Fluids and Electrolytes, (eds J. P. Kokkoand R. L. Tannen), W. B. Saunders, Philadelphia, PA, pp. 398–471.
Laureno, R. and Karp, B. I. (1988) Pontine and extrapontine myelinolysisfollowing rapid correction of hyponatraemia. Lancet, i, 1439–1441.
Lazrove, S., Waxman, K., Shippy, C. et al. (1980) Hemodynamic, blood volumeand oxygen transport responses to albumin and hydroxyethyl starchinfusions in critically ill postoperative patients. Critical Care Medicine, 8, 302.
Leaf, A. (1974) Hyponatraemia. Lancet, i, 1119–1120.Levine, O. R., Mellins, R. B., Senior, R. M. et al. (1967) The application of
Starling’s law of capillary exchange to the lungs. Journal of ClinicalInvestigation, 46, 934.
Levison, M. and Trunkey, D. D. (1982) Initial assessment and resuscitationSurgical Clinics of North America, 62, 9–16.
Lohr, J. W., McReynolds, J. and Grimaldi, T. (1988) Effect of acute and chronichypernatraemia on myoinositol and sorbitol concentration in rat brain andkidney. Life Science, 271–276.
Lowe, R. J., Moss, G. S., Jilek, J. et al. (1979) Crystalloid vs colloid in theetiology of pulmonary failure after trauma. A randomized trial in man.Critical Care Medicine, 7, 107.
Lowell, J. A., Schifferdecker, C., Driscoll, D. F. et al. (1990) Postoperative fluidoverload: Not a benign problem. Critical Care Medicine, 18(Suppl.), S193.
Lundy, E. F., Kuhn, J. E., Kwon, J. M. et al. (1987) Infusion of 5% dextroseincreases mortality and morbidity following six minutes of cardiac arrest inresuscitated dogs. Journal of Critical Care, 2, 4–14.
McMahon, M. M., Farnell, M. B. and Murray, M. J. (1993) Nutritional supportof critically ill patients. Mayo Clinic Proceedings, 68, 911–920.
Mangalore, P. P. and Hunt, T. K. (1972) Effect of varying oxygen tensions onhealing open wounds. Surgery, Gynecology, and Obstetrics, 135, 756.
Martinez-Maldonado, M. (1980) Inappropriate antidiuretic hormone secretionof unknown origin. Kidney International, 17, 554–567.
Mattox, K. L., Maningas, P. A., Moore, E. E. et al. (1991) Pre-hospitalhypertonic saline/dextran infusion for post-traumatic hypotension. Annalsof Surgery, 213, 482.
Messmer, K. (1975) Hemodilution. Surgical Clinics of North America, 55, 659.Meye, B. J., Meyer, A. and Guyton, A. (1968) Interstitial fluid pressure. Clinical
Research, 22, 263.Minnear, F. L., Kite, C., Hill, L. A. and Hoyte, Z. (1987) Endothelial injury and
pulmonary congestion characterize neurogenic pulmonary oedema inrabbits. Journal of Applied Physiology, 63(1), 335–341.
Mishle, J. (1984) Synthetic plasma volume expanders – their pharmacology,safety and clinical efficacy. Clinical Haematology, 13, 75.
Modig, J. (1986) Effectiveness of dextran 70 versus Ringer’s acetate intraumatic shock and adult respiratory distress syndrome. Critical CareMedicine, 14, 454.

330 FLUID, ELECTROLYTE AND METABOLIC MANAGEMENT
Monafo, W. W., Halverson, J. D. and Schechtman, K. (1984) The role ofconcentrated sodium solutions in the resuscitation of patients with severeburns. Surgery, 95, 129.
Moore, F. D., Dagher, F. J., Boyden, C. M. et al. (1966) Hemorrhage in normalman. 1. Distribution and dispersal of saline infusions following acuteblood loss: clinical kinetics of blood volume support. Annals of Surgery,163, 485.
Morissette, M. P. (1977) Colloid osmotic pressure: its measurement andclinical value. Canadian Medical Association Journal, 116, 897.
Morse, M. L., Milstein, J. M., Haas, J. E. et al. (1985) Effect of hydration onexperimentally induced cerebral edema. Critical Care Medicine, 13(7), 563.
Moss, G. S. and Gould, S. A. (1988) Plasma expanders. American Journal ofSurgery, 155, 425–434.
Moss, G. S., Lowe, R. J., Jilek, J. et al. (1981) Colloid or crystalloid in theresuscitation of haemorrhagic shock: a controlled clinical trial. Surgery, 89,434.
Muizelaar, J. P., Wei, E. P., Kontos, H. A. and Becker, D. P. (1983) Mannitolcauses compensatory cerebral vasoconstriction and vasodilation inresponse to blood viscosity changes. Journal of Neurosurgery, 59, 822–828.
Mulinari, R. A., Gavras, I., Wang, Y. X. et al. (1990) Effects of a vasopressinantagonist with combined antipressor and antidiuretic activities in ratswith left ventricular dysfunction. Circulation, 81, 308–311.
Muzaffar, T. Z., Stalker, A. L., Bryce, W. A. J. et al. (1973) Quantitative studieson fibrin formation and effects of dextran. Bibliotheca Anatomica, 12, 340.
Needleman, P. and Greenwald, J. E. (1986) Atriopeptin: a cardiac hormoneintimately involved in fluid, electrolyte, and blood-pressure homeostasis.New England Journal of Medicine, 314, 828–834.
Nelson, P. B., Seif, S. M., Maroon, J. C. and Robinson, A. G. (1981)Hyponatraemia in intracranial disease. Perhaps not the syndrome ofinappropriate secretion of antidiuretic hormone (SIADH). Journal ofNeurosurgery, 55, 938–941.
Niinikoski, J., Hengan, C. and Hunt, T. K. (1972) Oxygen tensions in humanwounds. Journal of Surgical Research, 12, 1977.
Norenberg, M. D. and Papendick, R. E. (1984) Chronicity of hyponatraemia asa factor in experimental myelinolysis. Annals of Neurology, 15, 544–547.
Oh, M. S. and Carroll, H. J. (1992) Disorders of sodium metabolism:hypernatraemia and hyponatraemia. Critical Care Medicine, 20(1), 94–103.
Packer, M., Medina, M. and Yushak, M. (1984) Correction of dilutionalhyponatremia in severe chronic heart failure by converting-enzymeinhibition. Annals of Internal Medicine, 100, 782–789.
Pappenheimer, J. R. and Soto-Rivera, A. (1948) Effective osmotic pressure ofthe plasma proteins and other quantities associated with the capillarycirculation in the hind limbs of cats and dogs. American Journal of Physiology,152, 471.
Park, B. E., Meacham, W. F. and Netsky, N. G. (1976) Nonketotic hyper-glycaemic hyperosmolar coma: report of neurosurgical cases with a reviewof mechanisms and treatment. Journal of Neurosurgery, 44, 409–417.
Paymaster, N. J. (1976) Magnesium metabolism: a brief review. Annals of theRoyal College of Surgeons of England, 58, 309–314.
Peters, R. M. and Hargens, A. R. (1981) Protein vs electrolytes and all of theStarling forces. Archives of Surgery, 116, 1293.
Pirisino, R., Di Simplicio, P., Ignesti, G. et al. (1988) Sulfhydryl groups andperoxidase-like activity of albumin as scavenger of organic peroxides.Pharmacology Research Community, 20, 545.
Plum, F. and Posner, J. B. (1980) The Diagnosis of Stupor and Coma, 3rd edn, F.A. Davis, Philadelphia, PA.
Poole, G. V., Johnson, J. C., Prough, D. S. et al. (1986) Cerebral haemodynamicsafter haemorrhagic shock: effects of the type of resuscitation fluid. CriticalCare Medicine, 14, 629.
Poole, G. V., Miller, J. D., Agnew, S. G. and Griswold, J. A. (1992) Lowerextremity fracture fixation in head injured patients. Journal of Trauma, 32,654–659.
Price, D. J. (1992) The intensive care of head-injured patients, in Care of theCritically Ill Patient, 2nd edn, (eds J. Tinker and W. M. Zapol), Springer-Verlag, London, pp. 831–872.
Price, D. J. E. and Murray, A. (1972) The influence of hypoxia and hypotensionon recovery from head injury. Injury, 3, 218–224.
Prough, D. S., Johnson, J. C., Poole, G. V. et al. (1985) Effects on intracranialpressure of resuscitation from haemorrhagic shock with hypertonic salineversus lactated Ringer’s solution. Critical Care Medicine, 13, 407.
Prough, D. S., Johnson, J. C., Stump, D. A. et al. (1986) Effects of hypertonicsaline versus lactated Ringer’s solution on cerebral oxygen transportduring resuscitation from hemorrhagic shock. Journal of Neurosurgery, 64,627.
Puri, V. K., Howard, M., Pidipaty, B. B. et al. (1983) Resuscitation inhypovolemia and shock: a prospective study of hydroxyethyl starch andalbumin. Critical Care Medicine, 11, 518.
Rackow, E. C., Falk, J. L., Fein, I. A. et al. (1983) Fluid resuscitation incirculatory shock: a comparison of the cardiorespiratory effects of albumin,hetastarch and saline solutions in patients with hypovolaemic and septicshock. Critical Care Medicine, 11, 839.
Rackow, E. C., Fein, I. A. and Leppo, J. (1977) Colloid osmotic pressure as aprognostic indicator of pulmonary oedema and mortality in the critically ill.Chest, 72, 709.
Rainey, T. G. and English, J. F. (1988) Pharmacology of colloids andcrystalloids, in The Pharmacologic Approach to the Critically Ill Patient, (ed. B.Chernow), Williams & Wilkins, Baltimore, MD, p. 219.
Ramming, S., Shackford, S. R., Zhuang, J. and Schmoker, J. D. (1994) Therelationship of fluid balance and sodium administration to cerebral oedemaformation and intracranial pressure in a porcine model of brain injury.Journal of Trauma, 37, 705–713.
Reed, L. L., Manglano, R., Martin, M. et al. (1991a) The effect of hypertonicsaline resuscitation on bacterial translocation after haemorrhagic shock inrats. Surgery, 110, 685.
Reed, R. L., Johnston, T. D., Chen, Y. et al. (1991b) Hypertonic saline altersplasma clotting times and platelet aggregation. Journal of Trauma, 31, 8.
Ring, J. and Messmer, K. (1977) Incidence and severity of anaphylactoidreactions to colloid volume substitutes. Lancet, i, 466.
Roberts, P., Pollay, M., Engles, L. et al. (1987) Effect on intracranial pressure offurosemide combined with varying doses and administration of mannitol.Journal of Neurosurgery, 66, 440–446.
Robertson, G. L. (1989) Syndrome of inappropriate antidiuresis. New EnglandJournal of Medicine, 321, 538–539.
Robin, A. P., Ing, T. S., Lancaster, G. A. et al. (1979) Hyperglycemia-inducedhyponatremia: a fresh look. Clinical Chemistry, 25, 496–497.
Robinson, A. G. (1985) Disorders of antidiuretic hormone secretion. Clinics inEndocrinology and Metabolism, 14, 55–88.
Ross, E. J. and Christie, S. B. M. (1969) Hypernatremia. Medicine, 48,441–472.
Roth, E., Lax, L. C. and Malone, J. V. (1969) Ringer’s lactate solution andextracellular fluid volume in the surgical patient: a critical analysis. Annalsof Surgery, 169, 149.
Salem, M., Munoz, R. and Chernow, B. (1991) Hypomagnesemia in criticalillness: a common and clinically important problem. Critical Care Clinics,7(1), 225–252.
Samson, W. K. (1987) Atrial natriuretic factor and the central nervous system.Endocrinology and Metabolism Clinics of North America, 16, 145–161.
Scheinkestel, C. D., Tuxen, D. V., Cade, J. F. and Shann, F. A. (1989) Fluidmanagement of shock in critically ill patients. Medical Journal of Australia,150, 508–517.
Schultze, R. G. and Nissenson, A. R. (1980) Potassium: physiology and patho-physiology, in Clinical Disorders of Fluid And Electrolyte Metabolism, 3rd edn,(eds M. H. Maxwell and C. R. Kleeman), McGraw-Hill, New York, pp.113–143.
Seckl, J. and Dunger, D. (1989) Postoperative diabetes insipidus. BritishMedical Journal, 298, 2–3.
Seckl, J. R., Dunger, D. B. and Lightman, S. L. (1987) Neurohypophysealfunction during early post- operative diabetes insipidus. Brain, 110,737–746.
Serkes, K. D. and Lang, S. (1966) Changes in extracellular fluid volume afterhemorrhage and tourniquet trauma. Surgery Forum, 17, 58.
Shackford, S. R. (1987) Fluid resuscitation of the trauma victim, in TraumaProblems in Critical Care, (eds S. R. Shackford and A. Perel), J. B. Lippincott,Philadelphia, PA, vol. 1 pp. 576–587.
Shapira, Y., Artae, A. A., Qassam, N. et al. (1995) Brain oedema and neurologicstatus with rapid infusion of 0.9% saline or 5% dextrose after head trauma.Journal of Neurosurgical Anesthesia, 7,17–25.
Shippy, C. R., Appel, P. L. and Shoemaker, W. C. (1984) Reliability of clinicalmonitoring to assess blood volume in critically ill patients. Critical CareMedicine, 12, 107.
Shires, G. T. (1965) The role of sodium containing solution in the treatment ofoligemic shock. Surgical Clinics of North America, 45, 365.
Shires, G. T. and Canizaro, P. C. (1973) Fluid resuscitation in the severelyinjured. Surgical Clinics of North America, 53, 1341.
Shires, G. T., Williams, J. and Brown, F. (1960) Simultaneous measurement ofplasma volume, extracellular fluid volume and red blood cell mass in manutilizing I131, S35, O4, Cr51. Journal of Laboratory and Clinical Medicine, 55,776.
Shires, T., Coln, D., Carrico, J. et al. (1964) Fluid therapy in haemorrhagicshock. Archives of Surgery, 88, 688.
Shires, G. T., Cunningham, J. N., Baker, C. R. F. et al. (1972) Alterations incellular membrane function during hemorrhagic shock in primates. Annalsof Surgery, 176, 288.
Shoemaker, W. C. (1976) Comparison of the relative effectiveness of wholeblood transfusions and various types of fluid therapy in resuscitation.Critical Care Medicine, 4, 71.
Shoemaker, W. C. (1982) Fluids and electrolytes in the adult, in Critical Care:State of the Art, (eds W. C. Shoemaker and W. L. Thompson), vol. 3, Societyof Critical Care Medicine, Fullerton, CA.
Shoemaker, W. C. and Czer, L. S. (1979) Evaluation of the biologic importanceof various hemodynamic and oxygen transport variables: which variablesshould be monitored in postoperative shock? Critical Care Medicine, 7,424.
Shoemaker, W. C. and Hauser, C. J. (1979) Critique of crystalloid versuscolloid therapy in shock and shock lung. Critical Care Medicine, 7, 117.
Shoemaker, W. C., Schluchter, M., Hopkins, J. A. et al. (1981) Comparison ofthe relative effectiveness of colloids and crystalloids in emergencyresuscitation. American Journal of Surgery, 142, 73.

REFERENCES 331
Shoemaker, W. C., Appel, P. L., Waxman, K. et al. (1982) Clinical trials ofsurvivors. 7 cardiorespiratory patterns as therapeutic goals in critically illpost-operative patients. Critical Care Medicine, 10, 398–403.
Shoemaker, W. C., Appel, P., Kram, H. et al. (1988) Prospective trial of supranormal values of survivors as therapeutic goals in high risk surgicalpatients. Chest, 94, 1176–1186.
Shucart, W. A. and Jackson, I. (1976) Management of diabetes insipidus inneurosurgical patients. Journal of Neurosurgery, 44, 65–71.
Siegel, J. H., Gens, D. R., Mamantov, T. et al. (1991) Effect of associated injuriesand blood volume replacement on death, rehabilitation needs, anddisability in blunt traumatic brain injury. Critical Care Medicine, 19,1252–1265.
Sinnatamby, C., Edwards, C. R. W., Kitau, M. and Irving, M. H. (1974)Antidiuretic hormone response to high and conservative fluid regimens inpatients undergoing operation. Surgery, Gynecology, and Obstetrics, 139,715–719.
Slutsky, A. S. (1993) American College of Chest Physicians ConsensusConference. Mechanical ventilation. Chest, 104, 1833–59.
Smith, S. D., Cone, J. B., Bowser, B. H. et al. (1982) Cerebral edema followingacute haemorrhage in a murine model: the role of crystalloid resuscitation.Journal of Trauma, 22(7), 588.
Solomon, S. M. and Kirby, D. F. (1990) The refeeding syndrome: a review.Journal of Parenteral and Enteral Nutrition, 14, 90–97.
Starling, E. H. (1896) On the absorption of fluids from the connective tissuespaces. Journal of Physiology (London), 9, 312.
Steinbok, P. and Thompson, G. B. (1978) Metabolic disturbances after headinjury: Abnormalities of sodium and water balance with special reference tothe effects of alcohol intoxication. Neurosurgery, 3(1), 9–15.
Sterns, R. H. (1987) Severe symptomatic hyponatraemia: treatment andoutcome. Annals of Internal Medicine, 107, 656–664.
Sterns, R. H., Cox, M., Felig, P. U. et al. (1981) Internal potassium balance andthe control of the plasma potassium concentration. Medicine, 60, 339–354.
Sterns, R. H., Riggs, J. E. and Schochet, S. S. Jr (1986) Osmotic demyelinationsyndrome following correction of hyponatraemia. New England Journal ofMedicine, 314, 1535.
Stocker, R., Glazer, A. N. and Ames, B. N. (1987) Antioxidant activity ofalbumin-bound bilirubin. Proceedings of the National Academy of Sciences ofthe USA, 84, 5918.
Stockigt, J. R. (1977) Potassium metabolism. Anaesthesia and Intensive Care, 5,317–325.
Sutin, K. M., Ruskin, K. J. and Kaufman, B. S. (1992) Intravenous fluid therapyin neurologic injury, in Critical Care Clinics vol. 8(2), (ed. B. S. Kaufman), W.B. Saunders, Philadelphia, PA, pp. 367–408.
Tannen, R. L. (1986) Potassium disorders, in Fluids and Electrolytes, (eds J. P.Kokko and R. L. Tannen), W. B. Saunders, Philadelphia, PA, pp.150–2228.
Taylor, A. E., Parker, J. C., Kvietys, P. R. et al. (1982) The pulmonaryinterstitium in capillary exchange. Annals of the New York Academy of Science,384, 146.
Tormey, W. P. (1990) Central pontine myelinolysis and changes in serumsodium. Lancet, 335, 1169.
Tranbaugh, R. F. and Lewis, F. R. (1985) Crystalloid fluid, in Controversies inTrauma Management (Clinics in Emergency Medicine), (eds R. N. Dailey andM. Callaham), Churchill Livingstone, pp. 121–133.
Vassar, M. J., Perry, C. A., Gannaway, W. L. et al. (1991) 7.5% sodium chloride/dextran for resuscitation of trauma patients undergoing helicopter trans-port. Archives of Surgery, 126, 1065.
Velanovich, V. (1989) Crystalloid versus colloid fluid resuscitation: a meta-analysis of mortality. Surgery, 105, 65.
Verney, E. B. (1954) Water diuresis (John Malet Purser Lecture). Irish Journal ofMedical Science, 345–377.
Virgilio, R. W., Smith, D. E. and Zarins, C. K. (1979) Balanced electrolytesolutions: Experimental and clinical studies. Critical Care Medicine, 7, 98.
Virgilio, R. W., Rice, C. L., Smithe, D. E. et al. (1979) Crystalloid vs colloidresuscitation. Is one better? Surgery, 85, 129–139.
Voll, C. L. and Auer, R. N. (1988) The effect of post ischaemic blood glucoselevels on ischaemic brain damage in the rat. Annals of Neurology, 24,638–646.
Wasil, M., Halliwell, B., Hutchison, D. C. et al. (1987) The antioxidant action ofhuman extracellular fluids. Biochemistry Journal, 243, 219.
Wasterlain, C. G. and Posner, J. B. (1968) Cerebral oedema in waterintoxication: I. Clinical and chemical observations. Archives of Neurology, 19,71–78.
Weaver, D. W., Ledgerwood, A. M., Lucas, C. E. et al. (1978) Pulmonary effectsof albumin resuscitation for severe hypovolaemic shock. Surgery, 113, 387.
Weed, L. H. and McKibben, P. S. (1919a) Experimental alteration of brain bulk.American Journal of Physiology, 48, 531.
Weed, L. H. and McKibben, P. S. (1919b) Pressure changes in the cerebrospinalfluid following intravenous injection of solutions of various concentrations.American Journal of Physiology, 48(4):512.
Weil, M. H. and Henning, R. J. (1979) New concepts in the diagnosis and fluidtreatment of circulatory shock. Anesthesia and Analgesia, 58, 124.
Weil, M. H., Morissette, M., Michaels, S. et al. (1974) Routine plasma colloidosmotic pressure measurements. Critical Care Medicine, 2, 229.
Weil, M. H., Michaels, S., Puri, V. K. et al. (1981) The stat laboratory: facilitatingblood gas and biochemical measurements for the critically ill and injured.American Journal of Clinical Pathology, 76, 34.
Weisberg, L. S. (1989) Pseudohyponatraemia: a reappraisal. American Journal ofMedicine, 86, 315–318.
Weiss, H. J. (1967) The effect of clinical dextran on platelet aggregation,adhesion and ADP release in man: in vivo and in vitro studies. Journal ofLaboratory and Clinical Medicine, 69, 37.
Weissman, C. (1990) The metabolic response to stress: an overview andupdate. Anaesthesiology, 73, 308–327.
Whang, R., Oei, T. O., Aikawa, J. K. et al. (1984) Predictors of clinicalhypomagnesemia. Hypokalemia, hypophosphatemia, hyponatremia andhypocalcemia. Archives of Internal Medicine, 144, 1794–1796.
Williams, G. H. (1986) Hyporeninemic hypoaldosteronism. New EnglandJournal of Medicine, 1041–1042.
Wilmore, D. W., Smith, R. J., O’Dwyer, S. T. et al. (1988) The gut – a centralorgan following surgical stress. Surgery, 104, 917.
Wise, B. L. and Chater, N. (1962) The value of hypertonic mannitol solution indecreasing brain mass and lowering cerebrospinal fluid pressure. Journal ofNeurosurgery, 19, 1038–1040.
Wisner, D. K., Schuster, L. and Quinn, C. (1990) Hypertonic saline resuscita-tion of head injury: Effects on cerebral water content. Journal of Trauma, 30,75.
Wittmers, L. E., Bartlett, M. and Johnson, J. A. (1976) Estimation of capillarypermeability coefficient of inulin in various tissues of the rabbit. Micro-vascular Research, 11, 67.
Worthley, L. I. G. (1986) Hyperosmolar coma treated with intravenous sterilewater. A study of three cases. Archives of Internal Medicine, 146, 945–947.
Worthley, L. I. G., Guerin, M. and Pain, R. W. (1987) For calculating osmolality,the simplest formula is the best. Anaesthesia and Intensive Care, 15,199–202.
Worthley, L. I. G. and Thomas, P. D. (1986) Treatment of hyponatraemicseizures with intravenous, 29.2% saline. British Medical Journal, 292,168–170.
Wray, N. P. and Nicotra, M. B. (1978) Pathogenesis of neurogenic pulmonaryoedema. American Review of Respiratory Disease, 118, 783–786.
Young, B., Ott, L., Norton, J. et al. (1985) Metabolic and nutritional sequelae inthe nonsteroid-treated head injury patient. Neurosurgery, 17, 784–791.
Zaloga, G. P. (1991) Hypocalcaemic crisis. Critical Care Clinics, 7, 191–200.Zaloga, G. P. and Chernow, B. (1986) Hypocalcemia in critical illness. Journal
of the American Medical Association, 256(14), 1924–1929.Zaloga, G. P., Chernow, B. and Cook, D. (1985) Assessment of calcium
homeostasis in the critically ill patient: the diagnostic pitfalls of the McLeanHastings nomogram. Annals of Surgery, 202, 587–594.
Zaloga, G. P., Chernow, B., Cook, D. et al. (1984) The importance of measuredserum ionized calcium levels in critically ill patients. Critical Care Medicine,12(3), 236.
Zornow, M. H., Scheller, M. S., Todd, M. M. et al. (1988) Acute cerebral effectsof isotonic crystalloid and colloid solutions following cryogenic braininjury in the rabbit. Anesthesiology, 69, 180.
Zornow, M. H., Todd, M. M. and Moore, S. S. (1987) The acute cerebral effectsof changes in plasma osmolality and oncotic pressure. Anaesthesiology, 67,936.
Zwimpfer, T. J. and Moulton, R. J. (1993) Neurologic trauma concerns, inCritical Care Clinics, vol. 9(4), (eds D. P. Milzman, B. R. Boulanger and A.Rodriguez), W. B. Saunders, Philadelphia, PA, pp. 727–739.



















