
1-s2.0-S0141813010001200-main (1)
6
International Journal of Biological Macromolecules 47 (2010) 298–303 Contents lists available at ScienceDirect International Journal of Biological Macromolecules journal homepage: www.elsevier.com/locate/ijbiomac Homology modeling and docking study of xanthine oxidase of Arthrobacter sp. XL26 R.G. Bodade a , S.D. Beedkar a , A.V. Manwar b , C.N. Khobragade a,∗ a Biochemistry Research Laboratory, School of Life Sciences, Swami Ramanand Teerth Marathawada University, Nanded, Maharashtra 431606, India b Department of Microbiology, Dyanopasak College, Parbhani, Maharashtra 431402, India article info Article history: Received 24 March 2010 Received in revised form 10 April 2010 Accepted 12 April 2010 Available online 18 April 2010 Keywords: Homology modeling Arthrobacter sp. XL26 XO (xodB protein) Docking abstract Hyperuricemia is a condition of defective purine metabolism characterized with elevated serum uric acid (UA) level that further leads to gout and gouty nephrolithiasis disorders. Gout is a world wide distributed rheumatic disease comprises 1% of the total population and still is in increasing state. One of the factors contributing to overproduction of UA is the hydroxylation of xanthine catalyzed by xanthine oxidase (XO). In the present study, 3D modeling of Arthrobacter sp. XL26 (xodB) protein was performed by comparative modeling approach using Rhodobacter capsulatus XDH (PDB ID: 2W3sF) as template in SWISS-MODEL, Geno3D and MODELLER program server. The best model was selected based on overall stereochemical quality (Procheck, PROSA, GenThreader), energy minimized, refined and used for active site characterization in BioMed CAChe workspace. The enzyme–inhibitor interaction was studied by docking to screen the possible inhibitors and application of model in design and development of anti-gout agents. © 2010 Elsevier B.V. All rights reserved. 1. Introduction Xanthineoxidoreductase (XOR) is a complex metallo- flavoprotein that catalyzes the oxidative hydroxylation of purines, pyrimidines, pterins and aldehyde substrates. It is also a key enzyme of purine pathway and catalyzes the conversion of hypoxanthine to xanthine and xanthine to uric acid (UA) with concomitant production of hydrogen peroxide and superoxide anions. It exhibits in two alternate forms of a same gene product as xanthine dehydrogenase (XDH, EC 1.1.1.204) and xanthine oxidase (XO, EC 1.2.3.2) [1,2]. Structurally the enzyme is a homodimer of molecular weight 290,000 and each subunit of the enzyme contains one molybdo-pterin (MO-Pt) cofactor, two distinct iron–sulfur cen- ters (2Fe–2S) and one flavin adenine dinucleotide cofactor (FAD) [3]. The accumulation of UA is known to cause hyperuricaemia, gout and UA nephrolithiasis and hence, inhibitors of UA formation could be useful as therapeutic agents for theses disease [4]. In addition a large amount of superoxide anion generation by XO has been involved in the pathogenesis of inflammation, mutagenesis, cancer, ageing and ischemic reperfusion injury, therefore inhibitors of the generation and radical scavengers of superoxide anion are also useful for prevention of the oxidative damaged induced diseases [5]. Moreover, gout is also associated with other diseases like hypertension, hyperlipidation, diabetes mellitus, obesity and ∗ Corresponding author. E-mail address: [email protected] (C.N. Khobragade). cardiovascular diseases [6]. The enzyme amino acid (1330 AA) sequence is highly homologous among the rat, mouse and human enzymes with about 90% identity [7]. A well characterized prokary- otic XDH with similar activity to the eukaryotic counterparts is the enzyme isolated from the phototrophic purple bacterium Rhodobacter capsulatus. The amino acid sequence of RcXDH has a high degree of similarity to eukaryotic XDH/XO. Because of the high structural and sequence similarity of RcXDH and bXDH especially in the active site, the bacterial enzyme is a good model system for studying the mechanism and design of the new, more effective clinical inhibitors [8]. Allopurinol is the most common clinically used XO inhibitor prescribed for the treatment of gout. However its use sometimes limited by hypersensitivity problems, Stevens–Johnson syndrome, renal toxicity, fetal liver necrosis and because of its poor scavenging activities [9]. Although, new as well as previously known compounds have recently been discovered to inhibit XDH/XO activity, a clinically effective inhibitor has not been developed to treat hyperuricemia after allopurinol. Recently febuxostat has been approved by FAD for gout treatment [10]. XO of Arthrobacter sp. XL26 has been purified and well characterized [11]. The enzyme revealed to has a MW of 260,000 and consists of heterogeneous subunits xodA and xodB that showed homology with Acinetobater baumannii ATCC 17978 XDH. Two-dimensional structure models indicate its (2Fe–2S) and FAD binding domains locate in xodA, whereas Mo-binding and a/b hammer head-domain of XDH seat in xodB. The crystal structure data (1FiQ) revealed direct substrate binding and oxidation reaction, at the MO-Pt center or MO ion of the MOCO [12]. 0141-8130/$ – see front matter © 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.ijbiomac.2010.04.002
-
Upload
zahid-khan -
Category
Documents
-
view
23 -
download
1
Transcript of 1-s2.0-S0141813010001200-main (1)

Ho
Ra
b
a
ARRAA
KHAD
1
flpehcax(mot[gcabcoadl
0d
International Journal of Biological Macromolecules 47 (2010) 298–303
Contents lists available at ScienceDirect
International Journal of Biological Macromolecules
journa l homepage: www.e lsev ier .com/ locate / i jb iomac
omology modeling and docking study of xanthine oxidasef Arthrobacter sp. XL26
.G. Bodadea, S.D. Beedkara, A.V. Manwarb, C.N. Khobragadea,∗
Biochemistry Research Laboratory, School of Life Sciences, Swami Ramanand Teerth Marathawada University, Nanded, Maharashtra 431606, IndiaDepartment of Microbiology, Dyanopasak College, Parbhani, Maharashtra 431402, India
r t i c l e i n f o
rticle history:eceived 24 March 2010eceived in revised form 10 April 2010ccepted 12 April 2010vailable online 18 April 2010
a b s t r a c t
Hyperuricemia is a condition of defective purine metabolism characterized with elevated serum uricacid (UA) level that further leads to gout and gouty nephrolithiasis disorders. Gout is a world widedistributed rheumatic disease comprises 1% of the total population and still is in increasing state. One ofthe factors contributing to overproduction of UA is the hydroxylation of xanthine catalyzed by xanthine
eywords:omology modelingrthrobacter sp. XL26 XO (xodB protein)ocking
oxidase (XO). In the present study, 3D modeling of Arthrobacter sp. XL26 (xodB) protein was performedby comparative modeling approach using Rhodobacter capsulatus XDH (PDB ID: 2W3sF) as template inSWISS-MODEL, Geno3D and MODELLER program server. The best model was selected based on overallstereochemical quality (Procheck, PROSA, GenThreader), energy minimized, refined and used for activesite characterization in BioMed CAChe workspace. The enzyme–inhibitor interaction was studied by
sible i
docking to screen the posagents.. Introduction
Xanthineoxidoreductase (XOR) is a complex metallo-avoprotein that catalyzes the oxidative hydroxylation of purines,yrimidines, pterins and aldehyde substrates. It is also a keynzyme of purine pathway and catalyzes the conversion ofypoxanthine to xanthine and xanthine to uric acid (UA) withoncomitant production of hydrogen peroxide and superoxidenions. It exhibits in two alternate forms of a same gene product asanthine dehydrogenase (XDH, EC 1.1.1.204) and xanthine oxidaseXO, EC 1.2.3.2) [1,2]. Structurally the enzyme is a homodimer of
olecular weight 290,000 and each subunit of the enzyme containsne molybdo-pterin (MO-Pt) cofactor, two distinct iron–sulfur cen-ers (2Fe–2S) and one flavin adenine dinucleotide cofactor (FAD)3]. The accumulation of UA is known to cause hyperuricaemia,out and UA nephrolithiasis and hence, inhibitors of UA formationould be useful as therapeutic agents for theses disease [4]. Inddition a large amount of superoxide anion generation by XO haseen involved in the pathogenesis of inflammation, mutagenesis,ancer, ageing and ischemic reperfusion injury, therefore inhibitors
f the generation and radical scavengers of superoxide anion arelso useful for prevention of the oxidative damaged inducediseases [5]. Moreover, gout is also associated with other diseasesike hypertension, hyperlipidation, diabetes mellitus, obesity and
∗ Corresponding author.E-mail address: [email protected] (C.N. Khobragade).
141-8130/$ – see front matter © 2010 Elsevier B.V. All rights reserved.oi:10.1016/j.ijbiomac.2010.04.002
nhibitors and application of model in design and development of anti-gout
© 2010 Elsevier B.V. All rights reserved.
cardiovascular diseases [6]. The enzyme amino acid (1330 AA)sequence is highly homologous among the rat, mouse and humanenzymes with about 90% identity [7]. A well characterized prokary-otic XDH with similar activity to the eukaryotic counterparts isthe enzyme isolated from the phototrophic purple bacteriumRhodobacter capsulatus. The amino acid sequence of RcXDH hasa high degree of similarity to eukaryotic XDH/XO. Because ofthe high structural and sequence similarity of RcXDH and bXDHespecially in the active site, the bacterial enzyme is a good modelsystem for studying the mechanism and design of the new, moreeffective clinical inhibitors [8]. Allopurinol is the most commonclinically used XO inhibitor prescribed for the treatment of gout.However its use sometimes limited by hypersensitivity problems,Stevens–Johnson syndrome, renal toxicity, fetal liver necrosis andbecause of its poor scavenging activities [9]. Although, new as wellas previously known compounds have recently been discoveredto inhibit XDH/XO activity, a clinically effective inhibitor has notbeen developed to treat hyperuricemia after allopurinol. Recentlyfebuxostat has been approved by FAD for gout treatment [10]. XOof Arthrobacter sp. XL26 has been purified and well characterized[11]. The enzyme revealed to has a MW of 260,000 and consistsof heterogeneous subunits xodA and xodB that showed homologywith Acinetobater baumannii ATCC 17978 XDH. Two-dimensional
structure models indicate its (2Fe–2S) and FAD binding domainslocate in xodA, whereas Mo-binding and a/b hammer head-domainof XDH seat in xodB. The crystal structure data (1FiQ) revealeddirect substrate binding and oxidation reaction, at the MO-Ptcenter or MO ion of the MOCO [12].
f Biolo
ttmotowmats
XAhXapaif
2
2
MM[cs
2
fbtAsmAuay(acwaat
hEPaGg
2
c
R.G. Bodade et al. / International Journal o
Therefore, knowledge of the 3D structure of a protein is essentialo understand how protein performs its function. Protein struc-ures can be determined at high resolution by either experimental
ethods such as X-ray crystallography and nuclear magnetic res-nance (NMR) or computational analysis (i.e. using bioinformaticsools) [13]. In the absence of crystallographic structure, a varietyf advanced homology modeling methods have been developed,hich can provide reliable models of proteins that share 30% orore sequence identity with a known structure [14]. Such models
lso have proven useful during drug design projects and allowedhe taking of key decisions in compound optimization and chemicalystems [15].
In continuation of our research work [16–18] to screen novelOR inhibitors, we purified and characterized the XO from isolatedrthrobacter strain (data unpublished). In the present study, theomology model of heterogeneous subunit from Arthrobacter sp.L26 (xodB) model has been generated by a comparative modelingpproach using R. capsulatus XDH (PDB ID: 2W3sF) chain as tem-late. The constructed model was further validated by structurenalysis tools like PROSA and PROCHECK to analyze its structuralntegrity. The active site prediction and docking studies were per-ormed to confirm its application for inhibitor screening.
. Methods
.1. Software and hardware
Automated homology modeling was performed by using SWISS-ODEL [19], Geno3D [20] and MODELLER 7v7 (http://salilab.org).odel was evaluated by PROCHECK [21], PROSA [22] and Verify-3D
23]. Docking studies with BioMed CAChe workspace module wasarried out [24]. Interactive visualization and analysis of moleculartructures was carried out in Swiss PDB viewer [25].
.2. Sequence retrieval alignment and homology modeling
The 3D model of heterogeneous subunit of XO (xodB)rom Arthrobacter sp. XL26 was built by homology modelingased on high-resolution crystal structures of homologous pro-eins. The complete amino acid sequence of the target proteinrthrobacter sp. XL26 (xodB) was retrieved from NCBI proteinequence database (Accession No. GI: 157058375) in FASTA for-at (http://www.ncbi.nlm.nih.gov/Proteins/). The NCBI Basic Locallignment Search Tool (BLAST), for the sequence similarities wassed for searching the crystal structures of the closest homologuesvailable in the Brookhaven Protein Data Bank (PDB). The resultsielded by NCBI BLAST revealed XDH (2W3sF) from R. capsulatusPDB ID: 2W3S) with a resolution of 2.6 Å as a suitable templatend with an identity score of 51% and E value 0.0. The coordinates ofrystal structure of XDH (2W3sF) from R. capsulatus (PDB ID: 2W3S)ere used as template to build the model by pair-wise sequence
lignment using Clustal W software [26]. GenThreader the fullyutomatic fold recognition server was used for fold assignment inarget [27].
Homology modeling of xodB was performed by three automatedomology modeling programs: SWISS-MODEL, Geno3D and MOD-LLER using single template downloaded from the BrookhavenDB (http://www.rcsb.org/pdb/) 2W3sF by comparative modelingpproach [28]. The steepest descent energy minimization using theROMOS 96 force field was done to regularize the protein structureeometry [29].
.3. Model refinement
The model was refined by performing an optimize geometry cal-ulation in mechanics using augmented MM3 parameter at BioMed
gical Macromolecules 47 (2010) 298–303 299
CAChe/workspace module. CAChe molecular mechanics includesenergy terms for bond stretch, bond angle, dihedral angle, impropertorsion stretch, bond bend, van der waals electrostatics and hydro-gen bond interactions. The refined model was again subjectedto energy minimization using GROMOS and evaluated for qualityassessment.
2.4. Validation of the 3D structure
The validation for structure models obtained from the differentsoftware tools (SWISS-MODEL, Geno3D and MODELLER) was per-formed by inspection of the Psi/Phi Ramachandran plot obtainedfrom PROCHECK analysis. Out of these, the model constructed bySWISS-MODEL was finally chosen for further investigations as itpossessed the best geometry and energy profile. The PROSA test wasapplied for final model to check for energy criteria in comparisonwith the potential of mean force derived from a large set of knownprotein structures. Further, the compatibility of the model with itssequence was measured by Verify-3D. The root mean square devia-tion (RMSD) between the main-chain atom of model and templatewas calculated by structural superimposition of template (2W3sF)and predicted structure of xodB heterogeneous subunit for the reli-ability of the model performed using combinatorial extension ofpolypeptides.
2.5. Active site prediction
The active site prediction in the refined model was done byautomatic sequence alignment mode in BioMed CAChe workspace.Active site was located in template first (2W3sF) by selecting ligand(xanthine), embedded in a 5 Å shell of residues, water and HETs andsaved as active site pocket. Active site pocket is the surface of theprotein adjacent to the ligand and the surface that forms a pocketaround the ligand. The active site detection in the refined modelwas done by automatic sequence alignment mode in BioMed CACheworkspace. We aligned the sequences of the homology model withthe sequence of the homologous template (2W3sF) for which theactive site is known. We identified the active site residues in homol-ogy model from the alignment. The gaps appear in the homologymodel and template was aligned in the sequences according tothe maximum scoring alignment using BLOSUM 50 substitutionmatrix in the Needlemham–Wunsch alignment algorithm [30–32].The matching residues in the homology model were discovered bymatch selection by choosing 2W3sF as the master sequence.
2.6. Docking ligand into homology model
The inhibitors were selected and refined for lowest energyrotamer by molecular mechanics procedure at BioMed CAChe 6.1.It was then docked over the homology model. CAChe automatesthe docking of ligand into the active site using a genetic algorithmwith a fast, simplified potential of mean force (PMF) [33]. PMFhas been demonstrated to show a significant correlation betweenexperimental binding affinities and its computed score for diverseprotein–ligand complexes [34–38].
3. Results and discussion
3.1. Homology modeling of xodB from Arthrobacter XL26
Homology modeling is usually the method of choice when a
clear relationship of homology between the sequence of targetprotein and at least one known structure is found. This approachwould give reasonable results based on the assumption that thetertiary structures of two proteins will be similar if their sequencesare related [39]. The absence of the three-dimensional structure
300 R.G. Bodade et al. / International Journal of Biological Macromolecules 47 (2010) 298–303
Fig. 1. Sequence alignment between XO of Arthrobacter sp. XL26 (xodB) protein and Rhodobacter capsulatus XDH (2W3sF chain) in Clustal W.
Table 1Possible template for 3D structure prediction of xodB protein.
Conf. Net score p-Value Pair E Solv E Aln score Aln Len Str Len Seq Len Alignment
CERT 355.668 6E−35 −1064.7 −19.2 1924.9 753 1298 784 1v97A0CERT 353.648 1E−34 −1078.9 −18.8 1908.2 754 1291 784 2e3tA0CERT 351.939 1E−34 −1099.4 −24.5 1869.9 759 760 784 2w3sB0CERT 338.834 3E−33 −1049.3 −14.1 1831 746 761 784 1rm6A0CERT 336.203 6E−33 −1034 −13.1 1818 760 786 784 1t3qB0CERT 330.582 2E−32 −947.4 −17.5 1787 755 795 784 1ffvB0CERT 321.011 2E−31 −1017.9 −10.7 1726 757 804 784 1n62B0CERT 272.214 2E−26 −810.8 −6.3 1463 720 906 784 1dgjA0CERT 269.7 3E−26 −862.9 −7.6 1427 723 907 784 1vlbA0CERT 188.687 4E−18 −547.6 −14.9 972 409 420 784 3hrdA0
fcxa(pBg
thm(ttatjg(ft
mtcMpps
in an unfavorable energy environment, which can be ignored inoverall consideration of the model structure. GROMOS has rep-resented the Y-axis of the plot, showing energy for each aminoacid of the protein chain in the form of red (unfavorable region)
CERT 134.254 1E−12 −435.6 3.6CERT 114.465 1E−10 201.2 25.5CERT 75.258 1E−06 270.9 40.8CERT 64.843 0.00001 87.8 16.7
or Arthrobacter xanthine oxidase (XO) in PDB prompted us toonstruct its homology model. The sequence of Arthrobacter sp.anthine oxidase was searched in NCBI site giving only two resultss Arthrobacter sp. XL26 (GI: 157092381) and Arthrobacter sp. XL26xodB) protein (GI: 157058375). The complete sequence of both theossible targets was downloaded in FASTA format and the proteinLAST for each complete protein sequence was executed, a hit wasiven <30% similarity with the target protein.
Among the XO/XDH with known three-dimensional structures,he 2W3sF XDH of the R. capsulatus (PDB ID: 2W3S) showed theighest sequence identity with xodB from Arthrobacter XL26. Pri-ary sequence alignment showed that target (xodB) and template
2W3sF) share 51% identity with E value 0.0 as compared to otheremplates. At this level of sequence identity, it is good enougho use crystallographic structure of 2W3sF from R. capsulatus as
template, in order to obtain high quality alignment for struc-ure prediction by homology modeling (Fig. 1). The neighboringoining plot for template and target also showed the similar ori-in at equal distance (23.19), confirming the structural homologyFig. 2). GeneThreader was used to recognize the fold assignmentor 3D structure prediction of Arthrobacter sp. XO (xodB) giving bestemplate score for 2W3sF as per Table 1.
The three-dimensional structure provides valuable insight intoolecular function and also enables the analyses of its interac-
ions with suitable substrates or inhibitors. Three models were
onstructed by different protein prediction tools: Geno3D, SWISS-ODEL and MODELLER. The predicted models were checked forsi and phi torsion angles using the Ramachandran plots. A com-arison of the results obtained from the different software tools,hows that one of the model generated by SWISS-MODEL is more
702 328 330 784 3hrdB0792 442 450 784 2w3sA0575 552 574 784 3c8yA0403 338 340 784 3i99A0
acceptable in comparison to those generated by Geno3D, and MOD-ELLER. Like 2W3sF crystal structure, the xodB homology model ispredominantly �-helical in nature with �-sheets as shown in Fig. 3.
3.2. Validation of the predicted structure
The possible applications of protein models depend largely onthe quality of models. The model was evaluated by the tools avail-able at SWISS-MODEL. The ANOLEA results represent the Y-axisof the plot, the energy for each amino acid of the protein chain.Negative energy values (in green) represents the favorable energyenvironment whereas the values (in red) unfavorable energy envi-ronment for a given amino acid. There are few amino acids lying
Fig. 2. Neighboring joining plot of Target (xodB) and Template (2W3sF) sequence.
R.G. Bodade et al. / International Journal of Biological Macromolecules 47 (2010) 298–303 301
Fig. 3. Refined structure of homology model of xodB. The �-helix is represented bydov
ot(Qnsmrwra(ir
ark pink spirals, �-sheet by yellow arrows and white are turns. (For interpretationf the references to color in this figure legend, the reader is referred to the webersion of the article.)
r green (favorable region) colorization. The force filed energy forhe overall structure is −1045.90 (before refinement) and −1072.16after refinement), confirm the homology model experimented.umean score is a composite score consisting of a linear combi-ation of six terms, which helps to estimate the quality of proteintructure model, in the form of a total Qumean score value. Theodel Qumean score obtained for the model (0.71) is within the
eliability (0–1). The overall stereochemical quality of the modelas assessed by Procheck. The Ramachandran plot showed 94.1%
esidues in most favorable region, 4.9% in allowed region, 1.1% in
dditional and disallowed region as compared to 2W3sF template94%, 5.0% and 0.9%), respectively. The results revealed that major-ty of the amino acids are in a phi–psi distribution consistent withight handed �-helix and reliable to be good quality model (Fig. 4).Fig. 4. Ramachandran plot of xodB homology model.
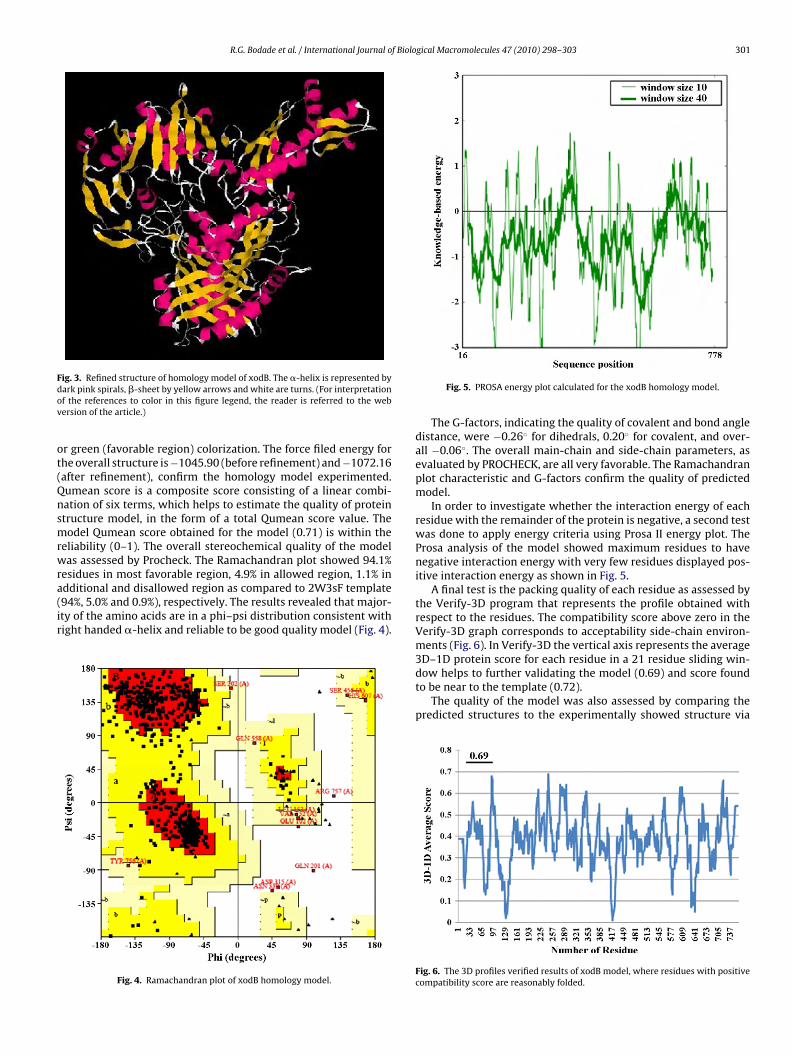
Fig. 5. PROSA energy plot calculated for the xodB homology model.
The G-factors, indicating the quality of covalent and bond angledistance, were −0.26◦ for dihedrals, 0.20◦ for covalent, and over-all −0.06◦. The overall main-chain and side-chain parameters, asevaluated by PROCHECK, are all very favorable. The Ramachandranplot characteristic and G-factors confirm the quality of predictedmodel.
In order to investigate whether the interaction energy of eachresidue with the remainder of the protein is negative, a second testwas done to apply energy criteria using Prosa II energy plot. TheProsa analysis of the model showed maximum residues to havenegative interaction energy with very few residues displayed pos-itive interaction energy as shown in Fig. 5.
A final test is the packing quality of each residue as assessed bythe Verify-3D program that represents the profile obtained withrespect to the residues. The compatibility score above zero in theVerify-3D graph corresponds to acceptability side-chain environ-ments (Fig. 6). In Verify-3D the vertical axis represents the average3D–1D protein score for each residue in a 21 residue sliding win-
dow helps to further validating the model (0.69) and score foundto be near to the template (0.72).The quality of the model was also assessed by comparing thepredicted structures to the experimentally showed structure via
Fig. 6. The 3D profiles verified results of xodB model, where residues with positivecompatibility score are reasonably folded.

302 R.G. Bodade et al. / International Journal of Biological Macromolecules 47 (2010) 298–303
F2t
sqwdwqbgn
3
bIaalto
ig. 7. Superimposed backbones of xodB homology model (blue) and templateW3sF (pink). (For interpretation of the references to color in this figure legend,he reader is referred to the web version of the article.)
uperimposition and atoms RMSD assessment (Fig. 7). Conse-uently superimposition of the template with homology modelas executed by combinatorial extension of polypeptides. The RMSeviation of C � trace between homology structure and templateas 0.33 Å support that generated model is reasonably good and
uite similar to template. Hence, the final model, which proved toe well validated in terms of geometry and energy profiles, sug-ests that the model is good enough to be a starting point for ourext phase of docking studies.
.3. Active site prediction and docking study
The protein–ligand complexes give greater insights in structureased drug design, so a protein–ligand complex was developed.
t gives a more detailed and accurate picture of the interactions
nd structural complementarities between the ligand and thective site. The active site pocket of template revealed that theigand is highly embedded in hydrogen bond donor region ofhe protein. The protein–ligand complex in generated homol-gy model is developed by sequence alignment and matchFig. 9. Docking of inhibitors on active
Fig. 8. Active site (pink) for the xodB homology model. (For interpretation of thereferences to color in this figure legend, the reader is referred to the web version ofthe article.)
selection procedures. Assuming that the ligand binding modesare similar in the target and the template protein structure,the coordinates of ligand was transferred from 2W3sF crystalstructure. Comparative active site analysis of template (2W3sF)and homology model (xodB) shows highly conserved residuesGlu232 (in model Glu245), Leu 303(Leu 316), Pro306(Pro319),Arg310(Arg323), Ala340(A353), Phe344(Phe357), Ser458(Ser456),Phe459(Phe457), Thr460(Asn458), Leu461(Ala459),Leu464(Tyr462), Ala526(Ala524), Ala528(Ala526), Ala529(Ala527),Ser530(Ser528), Glu730(Glu733), MO778(MO778). The active siteresidues in homology model Asn458, Ala459 and Tyr462 are dif-ferent from those residues involved in the active site of template(2W3sF). The catalytic active site contains the MO (molybdenumcofactor), which is conserved in both models, this confirms itsimportance in biding with the inhibitor. Finally, docking studyof the model was performed in order to elucidate its structuraland functional relevance in terms of inhibitor binding. Allopurinoland flavone, the well-known inhibitors, were successively docked
on to the active site of homology model. Fig. 8 shows docking ofinhibitor on to the active site where the Ser456, Phe457, Asn458,Ala526, Ala527 and MO778 are directly involved in biding with theinhibitors. Table 2 shows the results of the docking experiments insite of xodB homology model.

R.G. Bodade et al. / International Journal of Biolo
Table 2Dock score of inhibitors.
Inhibitor/substrate Dock score
tawat
4
oTwrcsi
A
U
R
[[
[
[[[[
[
[
[
[
[
[[[
[[
[[[
[[[
[[[
[
Xanthine −95.471Flavone −82.224Allopurinol −84.520
erms of calculated free energy of binding. The substrate xanthines ligand gives highest score (−95.471), confirms its tight bindingith the template/target. The inhibitor flavone and allopurinol
lso gives good score −82.224 and −84.520, respectively, confirmshe model has application in drug discovery (Fig. 9).
. Conclusion
In the present work the 3D model of xodB was constructed inrder to accomplish its molecular modeling and docking studies.he model was validated and further used for docking analysisith some well-known inhibitors viz. flavone and allopurinol. The
esulting docking solutions were analyzed for binding pattern andonformational analysis study. Further work is concentrated oncreening of novel flavones using the same model to make a majormpact on anti-gout chemotherapy.
cknowledgment
The authors are thankful to Head, School of Life Sciences, SRTMniversity, Nanded, for providing necessary facilities.
eferences
[1] G.D. Vogels, C. Van Der Drift, Bacteriol. Rev. 40 (2) (1976) 403–468.[2] J.M. Pauff, H. Cao, R. Hille, J. Biol. Chem. 284 (2009) 8760–8767.[3] Y. Kuwabara, T. Nishino, K. Okamoto, T. Matsumura, B.T. Eger, E.F. Pai, T. Nishino,
PNAS 100 (14) (2003) 8170–8175.
[4] N. Masuoka, I. Kubo, Biochem. Biophys. Acta 1688 (2004) 245–249.[5] B. Halliwell, J.M.C. Gutteridge, C.E. Cross, J. Lab. Clin. Med. 119 (1992) 598–620.[6] H.K. Choi, G. Curhan, Curr. Opin. Rheumatol. 17 (2005) 341–345.[7] L. Ling, Free Radic. Biol. Med. 77 (222) (2001) 1–10.[8] J.J. Truglio, K. Theis, S. Leimkuhler, R. Rappa, K.V. Rajagopalan, C. Kisker, NSLSActive Rep. 2 (2001) 50–53.
[[
[
gical Macromolecules 47 (2010) 298–303 303
[9] Y. Wang, J.X. Zhu, L.D. Kong, C. Yang, C.H.K. Cheng, X. Zhang, Basic Clin. Phar-macol. Toxicol. 94 (2004) 232–237.
10] K.-H. Yu, Rec. Pat. Inflamm. Allergy Drug Discov. 1 (2007) 69–75.11] L. Zhong-qin, X. Xiao-ping, X. Xiao-hua, W. Wu, Chin. J. of Pharma. 38 (12) (2007)
842–845.12] C.-M. Lin, C.-S. Chen, C.-T. Chen, Y.-C. Liang, J.-K. Lin, Biochem. Biophys. Res.
Commun. 294 (2002) 167–172.13] J.M. Sasin, J.M. Bujnicki, Nucleic Acids Res. 32 (2004) 586–589.14] S.K. Burley, Nat. Struct. Biol. 7 (Suppl.) (2000) 932–938.15] M.C. Peitsch, Biochem. Soc. Trans. 24 (1996) 274–279.16] C.N. Khobragade, R.G. Bodade, M.S. Shinde, D.R. Jaju, R.B. Bhosle, B.S. Dawane,
J. Enzyme Inhib. Med. Chem. 23 (3) (2008) 341–346.17] C.N. Khobragade, R.G. Bodade, A.V. Manwar, Asian J. Res. Chem. 3 (1) (2010)
139–141.18] C.N. Khobragade, R.G. Bodade, B.S. Dawane, S. Konda, N.T. Khandare, Inhib. Med.
Chem., doi:10.3109/14756360903389849.19] T. Schwede, J. Kopp, N. Guex, M.C. Peitsch, Nucleic Acids Res. 31 (2003)
3381–3385.20] C. Combet, M. Jambon, G. Deléage, C. Geourjon, Bioinformatics 18 (2002)
213–214.21] R.A. Laskowaski, M.W. McArther, D.S. Moss, J.M. Thornton, J. Appl. Crystallogr.
26 (1993) 283–291.22] M.J. Sippl, Proteins 17 (1993) 355–362.23] D. Eisenberg, R. Luthy, J.U. Bowie, Methods Enzymol. 277 (1997) 396–404.24] BioMed CAChe 6.1 Molecular Modelling Programme Package, Fujitsu Limited,
Life Science and Material Science Division 9-3 Nakase 1-Chame, Avihama-kuChiba City, Chiba 261-8588, Japan, 2006.
25] N. Guex, M.C. Peitsch, Electrophoresis 18 (15) (1997) 2714–2723.26] J.D. Thompson, T.J. Gibson, F. Plewniak, F. Jeanmougin, D.G. Higgins, Nucleic
Acids Res. 25 (1997) 4876–4882.27] L.J. McGuffin, D.T. Jones, Bioinformatics 19 (7) (2003) 874–881.28] A. Sali, T.L. Blundell, J. Mol. Biol. 234 (1993) 779–815.29] M. Christen, P.H. Hünenberger, D. Bakowies, R. Baron, R. Bürgi, D.P. Geerke, T.N.
Heinz, M.A. Kastenholz, V. Kräutler, C. Oostenbrink, C. Peter, D. Trzesniak, W.F.van Gunsteren, J. Comput. Chem. 26 (2005) 1719–1751.
30] S.B. Needleman, C.D. Wunsch, J. Mol. Biol. 48 (1970) 443–453.31] O. Gotoh, J. Mol. Biol. 264 (1996) 823–828.32] R. Durbin, S.R. Eddy, A. Krough, G. Mitchinson, Biological sequence analysis,
Cambridge University Press, Cambridge, U.K., 1998.33] I. Muegge, Y.C. Mrtin, J. Med. Chem. 42 (1999) 791–804.34] I. Muegge, Med. Chem. Res. 9 (1998) 490–500.35] I. Muegge, Y. Martin, P.J. Hajduk, S.W. Fesik, J. Med. Chem. 42 (1999) 2498–
2503.36] S. Ha, R. Andreani, A. Robbins, I. Muegge, J. Comput. Aided Mol. Des. 14 (2000)
435–448.
37] I. Muegge, M. Rarey, Rev. Comput. Chem. 17 (2001) 1–160.38] W.D. Cornell, P. Cieplak, C.I. Bayly, I.R. Gould, K.M. Merz Jr., D.M. Forguson, D.C.Spellmeyer, T. Fox, J.W. Caldwell, P.A. Kollman, J. Am. Chem. Soc. 117 (1995)5179–5197.
39] R.T. Kroemer, S.W. Doughty, A.J. Robinson, W.G. Richards, Protein Eng. 6 (1996)493–498.


















