
1 Podstawowe funkcje programu Gaussview/Gaussian
90
©Jan Pyka, Zakład Chemii Teoretycznej UAM 1 1 Podstawowe funkcje programu Gaussview/Gaussian Wszyscy chemicy posługują się modelami. Początkujący studenci używają plastykowych modeli, które pomagają im zrozumieć i wyobrazić sobie budowę cząsteczek. W ostatnich latach zarówno studenci, jak i doświadczeni badacze w coraz większym stopniu wykorzystują do tego samego celu chemiczne programy rysujące. Większość z nich opiera się na zbiorach odpowiednio zdefiniowanych obiektów oraz praw, które przybliżają prawdziwe chemiczne molekuły i procesy. W podobny sposób c h e m i a o b l i c z e n i o w a symuluje numerycznie chemiczne struktury oraz reakcje, bazując całkowicie lub po części na fundamentalnych prawach fizyki. Dzięki niej chemicy mogą badać zjawiska chemiczne nie tylko eksperymentalnie w laboratoriach, ale i wykonując obliczenia na komputerach. Ważną zaletą takiego podejścia jest możliwość uzyskania informacji, których nie dają nam metody obserwacyjne, np. o krótko istniejących obiektach pośrednich lub stanach przejściowych. Chemia obliczeniowa jest zatem zarazem niezależną dziedziną nauki i ważnym uzupełnieniem badań eksperymentalnych. Metody chemii obliczeniowej poświęcone badaniom struktury molekuł i ich reaktywności można podzielić na metody należące do dziedzin m e c h a n i k i m o l e k u l a r n e j i t e o r i i s t r u k t u r y e l e k t r o n o w e j . Wszystkie one umożliwiają przeprowadzanie tych samych podstawowych typów obliczeń: Obliczanie energii molekuł o odpowiednie zdefiniowanych strukturach (przestrzenne uporządkowanie atomów lub jąder i elektronów). Niektóre z tych metod umożliwiają także przewidzenie właściwości związanych z energią. Przeprowadzanie optymalizacji geometrii cząsteczki. Obliczanie częstości drgań molekuł. W mechanice molekularnej do przewidywania struktur i właściwości cząsteczek wykorzystuje się prawa fizyki klasycznej. Jej metody dostępne są w licznych programach komputerowych, np. HyperChem, Quanta, Sybyi lub Alchemy. Metody należące ogólnie do teorii struktury elektronowej bazują na prawach mechaniki kwantowej, a nie fizyki klasycznej. Istnieją dwie główne klasy tych metod: Metody półempiryczne, np AM1, MINDO/3 i PM3, które są zaimplementowane w takich programach jak MOPAC, HyperChem i G a u s s i a n . Wykorzystuje się w nich parametry uzyskane z danych doświadczalnych, dzięki czemu obliczenia znacznie się upraszczają.
Transcript of 1 Podstawowe funkcje programu Gaussview/Gaussian

©Jan Pyka, Zakład Chemii Teoretycznej UAM
1
1 Podstawowe funkcje programu Gaussview/Gaussian Wszyscy chemicy posługują się modelami. Początkujący studenci używają plastykowych
modeli, które pomagają im zrozumieć i wyobrazić sobie budowę cząsteczek. W ostatnich
latach zarówno studenci, jak i doświadczeni badacze w coraz większym stopniu wykorzystują
do tego samego celu chemiczne programy rysujące. Większość z nich opiera się na zbiorach
odpowiednio zdefiniowanych obiektów oraz praw, które przybliżają prawdziwe chemiczne
molekuły i procesy.
W podobny sposób c h e m i a o b l i c z e n i o w a symuluje numerycznie chemiczne
struktury oraz reakcje, bazując całkowicie lub po części na fundamentalnych prawach fizyki.
Dzięki niej chemicy mogą badać zjawiska chemiczne nie tylko eksperymentalnie w
laboratoriach, ale i wykonując obliczenia na komputerach. Ważną zaletą takiego podejścia
jest możliwość uzyskania informacji, których nie dają nam metody obserwacyjne, np. o
krótko istniejących obiektach pośrednich lub stanach przejściowych. Chemia obliczeniowa
jest zatem zarazem niezależną dziedziną nauki i ważnym uzupełnieniem badań
eksperymentalnych.
Metody chemii obliczeniowej poświęcone badaniom struktury molekuł i ich reaktywności
można podzielić na metody należące do dziedzin m e c h a n i k i m o l e k u l a r n e j i
t e o r i i s t r u k t u r y e l e k t r o n o w e j . Wszystkie one umożliwiają przeprowadzanie
tych samych podstawowych typów obliczeń:
Obliczanie energii molekuł o odpowiednie zdefiniowanych strukturach
(przestrzenne uporządkowanie atomów lub jąder i elektronów). Niektóre z tych
metod umożliwiają także przewidzenie właściwości związanych z energią.
Przeprowadzanie optymalizacji geometrii cząsteczki.
Obliczanie częstości drgań molekuł.
W mechanice molekularnej do przewidywania struktur i właściwości cząsteczek
wykorzystuje się prawa fizyki klasycznej. Jej metody dostępne są w licznych programach
komputerowych, np. HyperChem, Quanta, Sybyi lub Alchemy.
Metody należące ogólnie do teorii struktury elektronowej bazują na prawach mechaniki
kwantowej, a nie fizyki klasycznej. Istnieją dwie główne klasy tych metod:
Metody półempiryczne, np AM1, MINDO/3 i PM3, które są zaimplementowane w
takich programach jak MOPAC, HyperChem i G a u s s i a n . Wykorzystuje się w
nich parametry uzyskane z danych doświadczalnych, dzięki czemu obliczenia
znacznie się upraszczają.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
2
Metody ab initio, w których podczas obliczeń – w przeciwieństwie do mechaniki
molekularnej lub metod półemiprycznych – nie wykorzystuje się żadnych
parametrów eksperymentalnych, bowiem oparte są wyłącznie na prawach
mechaniki kwantowej oraz małej liczbie stałych fizycznych, a mianowicie:
o prędkości światła
o masach i ładunkach elektronów oraz jąder atomowych
o stałej Plancka.
W programie ( a właściwie pakiecie programów) Gausian 03 dostępne wiele różnych
metod służących do obliczeń struktury elektronowej molekuł. My będziemy stosować
głównie metodę HF (Hartree-Focka)

©Jan Pyka, Zakład Chemii Teoretycznej UAM
3
2 GaussView GaussView jest graficznym interfejsem do programu Gaussian 03, umożliwiającym
konstruowanie cząsteczek w prosty, graficzny sposób, wprowadzenie odpowiednich
parametrów obliczeniowych oraz dzięki powiązaniu z programem Gaussian zarządzanie
obliczeniami. Po uruchomieniu programu GaussView otwierają się dwa okna:
Pierwsze z nich to okno programu GaussView, a drugie to okno widoku (View) o nazwie
G1:M1:V1 - New, w którym będziemy budować molekuły. W oknie programu domyślnie
(default) znajduje się tetraedryczny atomu węgla. Po kliknięcie myszką w dowolnym miejscu
okna widoku zostaje on skopiowany do niego, jak to pokazano na rysunku. Można utworzyć
wiele okien widoku, wybierając z menu okna głównego GaussView kolejne opcje File,
następnie New, a potem Create MolGroup, i do każdego z nich wprowadzać (kliknięciami
myszki) fragmenty cząsteczki z okna GaussView.
2.1 Ustawienia opcji wyświetlania i obracanie cząsteczki
Podczas konstruowania cząsteczki możemy ją obracać, powiększać oraz przesuwać. W
tym celu używamy przycisków myszy: Lewy przycisk myszy - pojedyncze kliknięcie dodaje
atom lub fragment, dłuższe przytrzymanie umożliwia obracanie cząsteczki. Środkowy
Okno główne
programu
GaussView
Okno widoku

©Jan Pyka, Zakład Chemii Teoretycznej UAM
4
przycisk myszy - kliknięcie i przytrzymanie przycisku umożliwia przesuwanie cząsteczki w
oknie. Prawy przycisk myszy dłuższe przytrzymanie klawisza i ruch myszy umożliwia
powiększanie i zmniejszanie cząsteczki, pojedyncze kliknięcie powoduje wyświetlenie
podręcznego menu, w którym możemy zaznaczyć opcje wyświetlania:
Center – ustawia cząsteczkę w centrum okna
Hydrogens – włącza pokazywanie atomów wodoru
Dummies – włącza pokazywanie dodatkowych atomów, jeśli są
Labels – włącza wyświetlanie numeru atomu
Symbols – dodaje symbole pierwiastków do wyświetlanego widoku
Bonds – pokazuje wiązania w cząsteczce
Cartesian Axes – pokazuje układ współrzędnych
Display Format – oferuje dodatkowe ustawienia opcji widoku cząsteczki
Uwaga: Obiekty w oknie widoku można także przesuwać, a także powiększać i
pomniejszać poprzez naciśnięcie lewego przycisku myszki i klawisza Shift lub Ctrl:
Shift – przesuwamy obiekt po oknie
Ctrl – obracamy obiekt, jednocześnie go powiększając lub pomniejszając
W sytuacji, gdy w oknie widoku są dwa obiekty i chcemy obrócić tylko jeden z nich,
wskazujemy go myszką, naciskamy jej lewy przycisk i klawisz Alt. Jeśli chcemy zbliżyć
jeden obiekt do drugiego, wskazujemy go myszką i naciskamy jej lewy przycisk oraz
kombinację klawiszy Shift + Alt lub Ctrl.
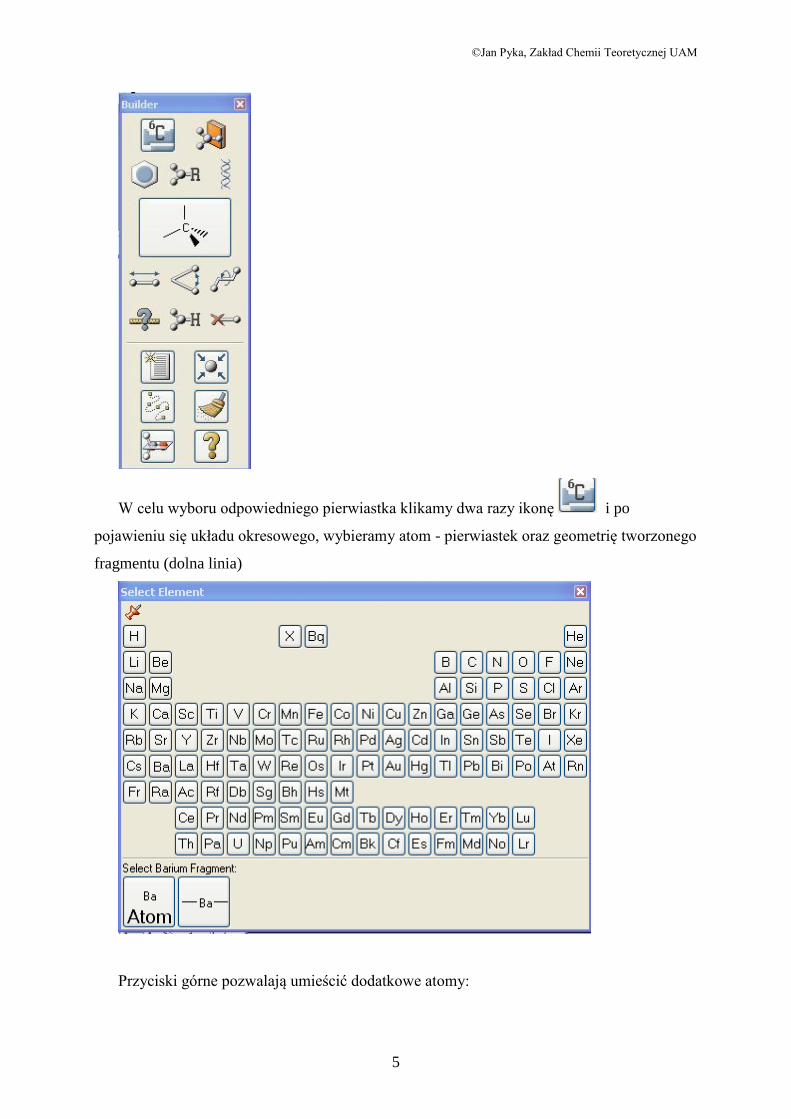
Konstruowanie zaczynamy od wyboru odpowiednich atomów, fragmentów lub
związków. W tym celu możemy posługiwać się ikonami z podstawowego okna programu lub
otworzyć dodatkowe okno z modułem Molecule Builder. Dokonujemy to przez zaznaczenie
w menu View opcji Builder (Konstruktor). Pojawia się nowe okno, z którego wybieramy
żądany element:
©Jan Pyka, Zakład Chemii Teoretycznej UAM
5
W celu wyboru odpowiedniego pierwiastka klikamy dwa razy ikonę i po
pojawieniu się układu okresowego, wybieramy atom - pierwiastek oraz geometrię tworzonego
fragmentu (dolna linia)
Przyciski górne pozwalają umieścić dodatkowe atomy:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
6
- dummy atom - umożliwia odnoszenie się do określonego punktu podczas
konstruowania molekuły w układzie kartezjańskim,
- ghost atom -umożliwia to poszerzenie bazy o dodatkowe orbitale wirtualnego
atomu.
Po wybraniu atomu oraz odpowiedniej geometrii fragmentu klikamy w oknie, w którym
tworzymy cząsteczkę i wstawiamy odpowiednim miejscu wybrany atom. Wybrana geometria
jest dopełniana wiązaniami z wodorem lub widoczna tylko w formie wiązań.
2.2 Wybór fragmentu
Aby wybrać odpowiedni fragment (pierścień) z podręcznego menu klikamy przycisk
. W nowym oknie, które się pojawi wybieramy odpowiedni fragment. Następnie poprzez
klikniecie w oknie konstruowania cząsteczki wstawiamy odpowiedni element.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
7
2.3 Wybór grupy.
Aby wybrać odpowiednią grupę klikamy na ikonę , a po pojawieniu się okna
wybieramy interesującą nas grupę i wstawiamy ją (poprzez kliknięcie) do okna, w którym
konstruujemy cząsteczkę.
2.4 Dodawanie pojedynczych aminokwasów lub nukleotydów
Aby w prosty sposób zbudować peptyd lub fragment łańcucha nukleotydowego
wykorzystujemy już zdefiniowaną bibliotekę aminokwasów oraz nukleotydów ukrytą pod
ikoną . Klikając dwa razy uzyskujemy w nowym oknie możliwość wyboru aminokwasu
lub nukleotydu, a także pozycji, jaką on zajmie w naszej cząsteczce.
2.5 Dodawanie oraz edycja własnych fragmentów cząsteczek do biblioteki

©Jan Pyka, Zakład Chemii Teoretycznej UAM
8
Chcąc dodać naszą własną cząsteczkę lub fragment do podręcznego zbioru po
skonstruowaniu cząsteczki użyjemy przycisku , w wyniku czego pojawi się okno
umożliwiające zapisanie dodawanego elementu.
2.6 Opcja „HOT” Atom.
Po wybraniu atomu lub fragmentu cząsteczki pojawia się w oknie głównym struktura
wybranego elementu z zaznaczonym „gorącym” atomem. Jest to atom, który zastępuje atom
w miejscu kliknięcia na cząsteczkę w oknie konstruowania.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
9
Na przykład dla wybranego atomu węgla o hybrydyzacji sp3, możemy ( klikając myszką
w odpowiednich miejscach) ustawić dwie pozycje „gorącego” atomu
W oknie, w którym konstruujemy cząsteczkę po wybraniu „HOT” atomu klikamy na
jeden z atomów wodoru wcześniej utworzonej cząsteczki metanu.
W zależności od zaznaczonego (gorącego) atomu uzyskujemy różne struktury cząsteczek.
©Jan Pyka, Zakład Chemii Teoretycznej UAM
10
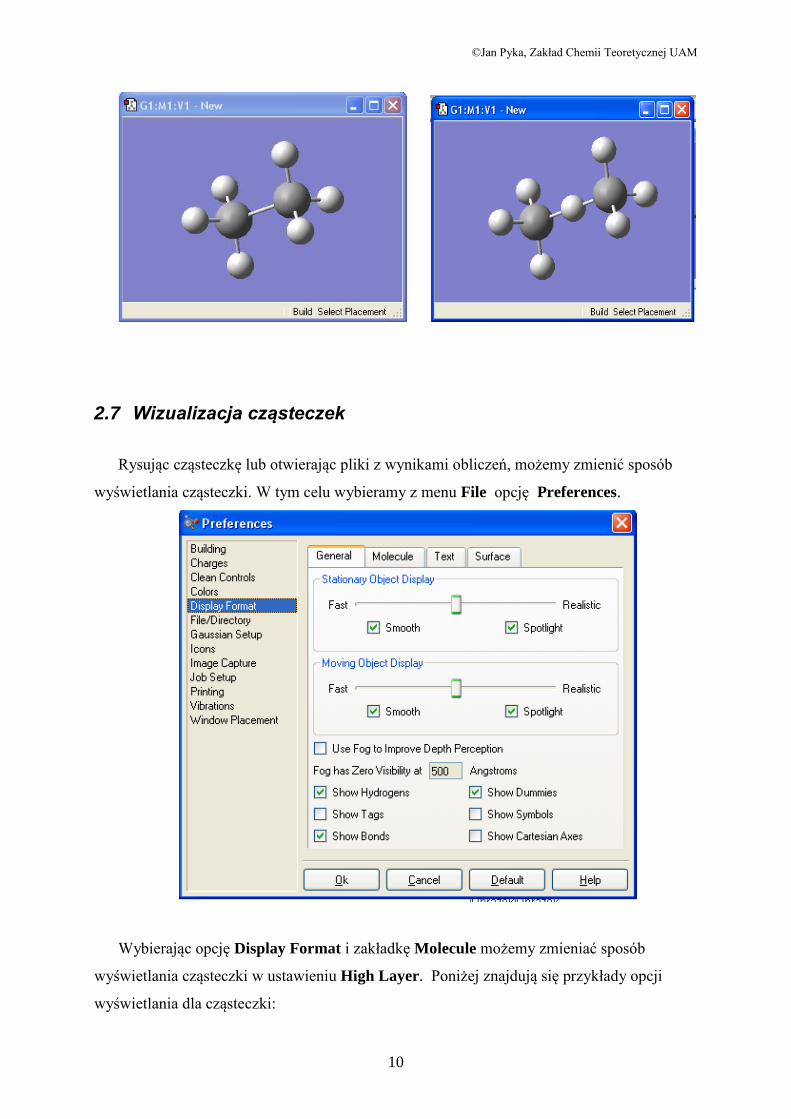
2.7 Wizualizacja cząsteczek
Rysując cząsteczkę lub otwierając pliki z wynikami obliczeń, możemy zmienić sposób
wyświetlania cząsteczki. W tym celu wybieramy z menu File opcję Preferences.
Wybierając opcję Display Format i zakładkę Molecule możemy zmieniać sposób
wyświetlania cząsteczki w ustawieniu High Layer. Poniżej znajdują się przykłady opcji
wyświetlania dla cząsteczki:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
11
Ball & Bond Type Ball & Stick
Tube
Twórcy programu GaussView położyli nacisk na prostotę oraz na szybkość wyświetlania
grafiki. Użytkownik może „oglądać wnętrze” molekuły za pomocą suwaka Z-clip . W innych
zakładkach tego okna można ustawić dodatkowe opcje wyświetlania, na przykład
pokazywanie atomów wodorów (zakładka General), czy etykiet (zakładka Text).
2.8 Zmiana odległości między atomami
Klikamy na ikonę a następnie na dwa atomy w cząsteczce, wówczas dla
cząsteczki np. etenu pojawi się okno z parametrami wiązania:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
12
W tym momencie możemy wybrać typ wiązania wyświetlanego oraz ustawić odległość
między atomami. Odległość ustawiamy, posługując się suwakiem lub bezpośrednio wpisując
konkretną wartość do okna tekstowego. Uwaga: nie stosujemy przecinka dziesiętnego, lecz
kropkę. W opcji Displacement decydujemy, czy przesuwamy pojedynczy atom nr [1], [2]
(Translate atom), czy też całe grupy (Translate group), lub zaznaczyć, że pozycja
określonego atomu jest stała (Fixed). Warto wiedzieć, że wybór typu wiązania ma tylko
znaczenie wizualne i nie wpływa na wynik obliczeń Gaussiana.
©Jan Pyka, Zakład Chemii Teoretycznej UAM
13
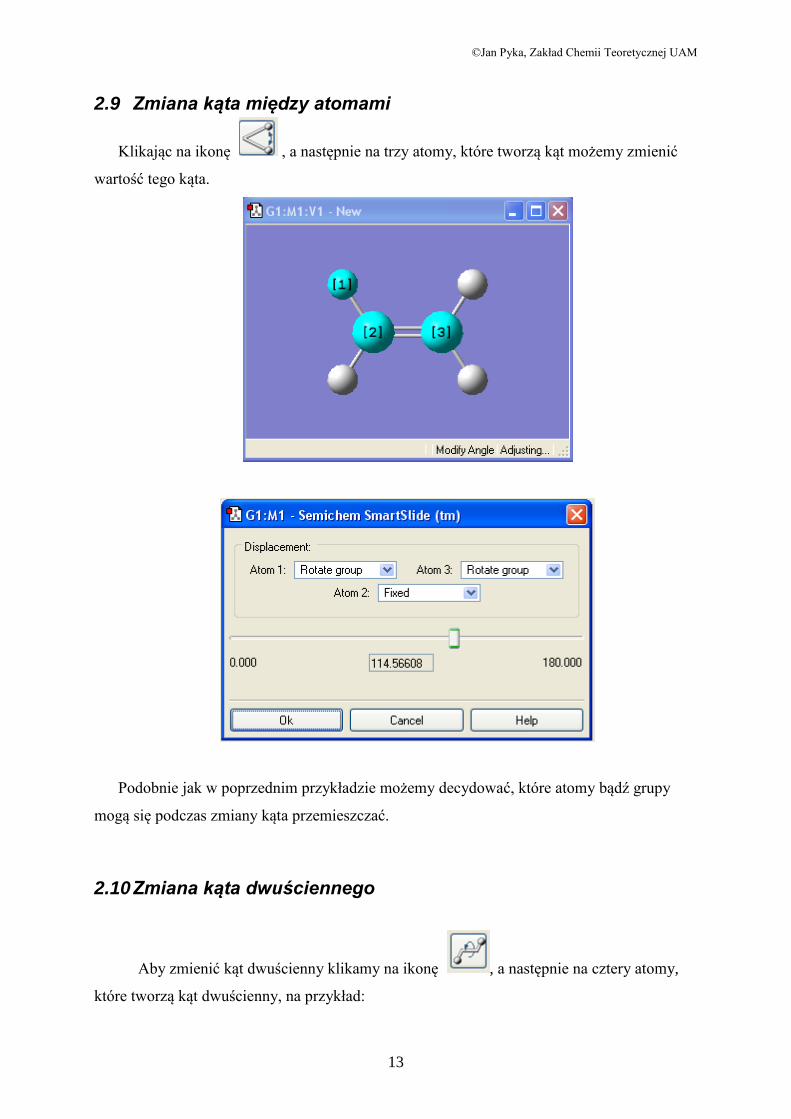
2.9 Zmiana kąta między atomami
Klikając na ikonę , a następnie na trzy atomy, które tworzą kąt możemy zmienić
wartość tego kąta.
Podobnie jak w poprzednim przykładzie możemy decydować, które atomy bądź grupy
mogą się podczas zmiany kąta przemieszczać.
2.10 Zmiana kąta dwuściennego
Aby zmienić kąt dwuścienny klikamy na ikonę , a następnie na cztery atomy,
które tworzą kąt dwuścienny, na przykład:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
14
Po zaznaczeniu czterech atomów otwiera się okno:
Możemy teraz ustawić wartość kąta w sposób podobny do ustawienia wiązania, na
przykład zmieniając wartość kąta dwuściennego na 90 stopni:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
15
2.11 Odczytywanie (a nie modyfikowanie) wartości parametrów geometrycznych
Przycisk Inquire umożliwia nam odczytanie bezpośrednio z okna widoku po
wybraniu interesujących nas atomów. Sprawdźmy, na przykład, jaki kąt tworzą atomy węgla
w benzenie:
Wartość kąta została wyświetlona w dolnej linii tego okna.
2.12 Usuwanie atomów oraz dodawanie atomów wodoru
W celu usunięcia jakiegoś atomu, klikamy na ikonę (Delete Atom) a następnie na
atom, który ma być usunięty. Aby dodać atom wodoru do wybranego atomu, klikamy na
ikonę (Add Valence) a następnie na wybrany atom.
2.13 Reszta przycisków z Konstruktora
Na dole okna Konstruktora (Builder) znajdują się jeszcze przyciski:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
16
– New: Tworzy nowy model i związane z nim okno
– Center: Ustawia molekułę w środku okna i zmieniaj jej rozmiar
– Rebond: Po naciśnięciu tego przycisku GaussView reidentyfikuje atomy, bazując
na algorytmie obliczającym odległości
– Clean: Dostosowanie geometrii molekuły, na podstawie zdefiniowanych
wcześniej zasad, do postaci, która lepiej odpowiada chemicznej intuicji. Wyniki są tylko
przybliżone i nie mają by idealne.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
17
3 Używanie różnych menu programu GaussView
Program jest wyposażony w pięć rozwijanych menu : File, Edit, View, Calculate i
Result. Dzięki zawartym w nich opcjom można kontrolować postać budowanych
modeli. Poniżej zostaną przedstawione niektóre z opcji pierwszego z tych menu;
pozostałe będziemy omawiać później, w miarę potrzeby.
3.1 Opcje menu File
Opcje menu File pozwalają tworzyć, modyfikować i zapisywać struktury chemiczne.
3.1.1 New: Tworzenie nowego modelu
Wybranie New powoduje utworzenie nowego modułu i związanego z nim okna widoku,
oraz ustawienie wszystkich opcji w oknach dialogowych zgodnie z ustawionymi
preferencjami. Warto zauważyć, że tę samą funkcję spełnia przycisk New w oknie
Konstruktora.
3.1.2 Open: Otwieranie plików.
Ta opcja pozwala otwierać pliki o różnych formatach wspieranych przez GaussView. Po
wybraniu Open otwiera się okno dialogowe Open File, typowe dla wszystkich programów
działających pod Windows. Między innymi widać w nim typy plików wykorzystywanych
przez GaussianView, między innymi:
Pliki wynikowe Gaussiana (.log lub .out)
Pliki danych wejściowych Gaussiana (.com lub .gjf)
Pliki typu checkpoint (.chk)
Pliki sformatowane typu checkpoint (.fch)
Pliki typu cube (.cub)
3.1.3 Save: zapisywanie plików
Dzięki tej opcji można zapisywać pliki w wybranym formacie

©Jan Pyka, Zakład Chemii Teoretycznej UAM
18
4 Budowanie molekuł: przykłady
Podane poniżej przykłady są ilustracją sposobów budowania cząsteczek chemicznych.
4.1 Pirydyna
Wybierz Pierścienie (Ring Fragment) w oknie Konstruktora.
Wybierz benzen
Powróć do okna widoku. Kliknij raz: pojawi się w nim molekuła benzenu
Kliknij (dwa razy) na tablicę pierwiastków (Element Fragment)
W tablicy wybierz atom azotu – bez hybrydyzacji
W oknie widoku kliknij na atom węgla, który chcesz zastąpić azotem
Jeśli pozostało jakieś wiązanie z wodorem, usuń je za pomocą przycisku Usuń
Atom (Delete Atom)
Naciśnij przycisk Rebond w oknie Konstruktora, a potem kliknij na przycisk
Clean. Zwykle należy robić to w tej właśnie kolejności
Tak oto zbudowaliśmy molekułę pirydyny.
4.2 Fenylopirydyna
W tym przykładzie zobaczymy, jak można zbudować cząsteczkę fenylopirydyny, bazując
istniejącym już modelu, w tym przypadku pirydynie.
Kontynuujemy zadanie z poprzedniego przykładu, czyli budowania pirydyny

©Jan Pyka, Zakład Chemii Teoretycznej UAM
19
Kliknij na Pierścienie w oknie Konstruktora. Wybierz pierścień fenylowy
W oknie widoku kliknij na ten atom wodoru pirydyny, który znajduje się naprzeciw
atomu azotu. Na ekranie pojawi się drugi pierścień
W oknie Konstruktora wybierz przycisk Kąt Dwuścienny (Dihedral). Upewnij
się, że zaznaczona jest opcja Rotate Groups
Wybierz atomy między pierścieniami tak, jak to widać na poniższym obrazku.
Wpisz w oknie tekstowym wartość dokładnie 90 stopni.
Kliknij OK.
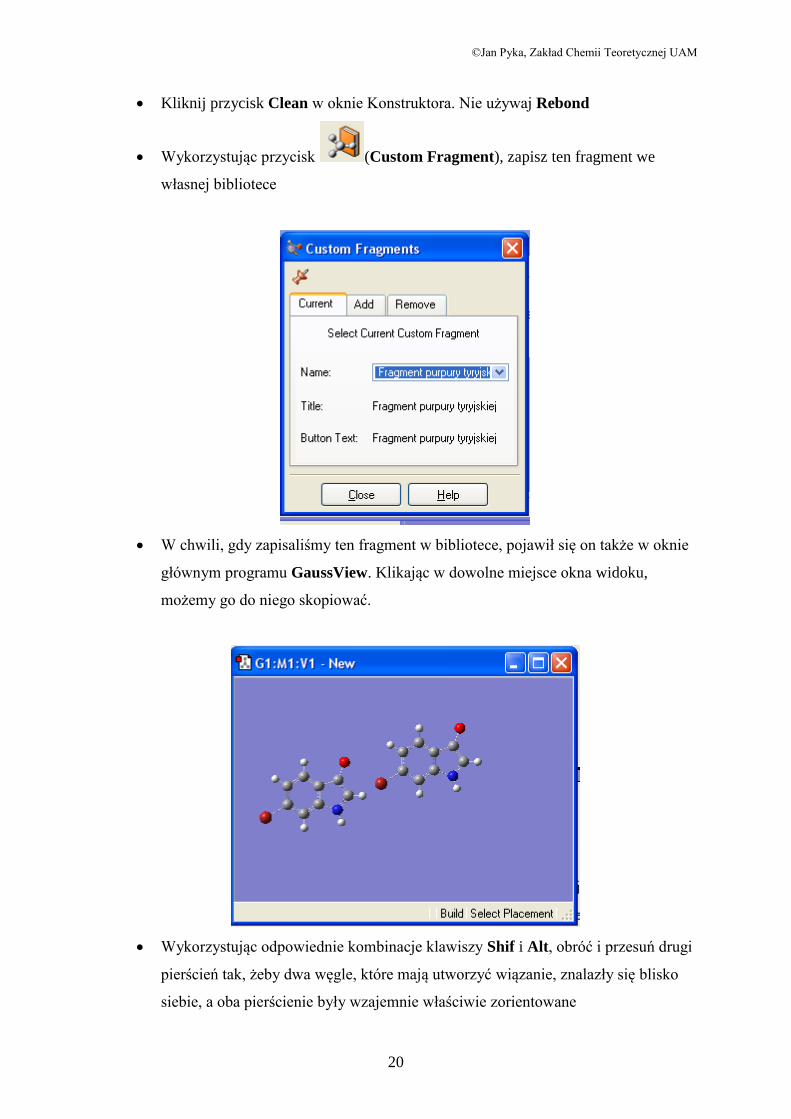
4.3 Purpura tyryjska
Utwórz nowe okno widoku
W oknie Pierścienie wybierz i kliknięciem skopiuj te pierścienie do okna
widoku
W tablicy pierwiastków wybierz tlen i wstaw go na miejsce odpowiedniego wodoru
w pierścieniu pięcioelementowym.
Zamień atom węgla znajdujący się naprzeciw tlenu na azot
Wykorzystując przycisk Add Valence dodaj wodór do azotu
Zamień atom wodoru znajdujący się na końcu pierścienia sześcioelementowego (po
tej samej stronie co azot) na brom.
©Jan Pyka, Zakład Chemii Teoretycznej UAM
20
Kliknij przycisk Clean w oknie Konstruktora. Nie używaj Rebond
Wykorzystując przycisk (Custom Fragment), zapisz ten fragment we
własnej bibliotece
W chwili, gdy zapisaliśmy ten fragment w bibliotece, pojawił się on także w oknie
głównym programu GaussView. Klikając w dowolne miejsce okna widoku,
możemy go do niego skopiować.
Wykorzystując odpowiednie kombinacje klawiszy Shif i Alt, obróć i przesuń drugi
pierścień tak, żeby dwa węgle, które mają utworzyć wiązanie, znalazły się blisko
siebie, a oba pierścienie były wzajemnie właściwie zorientowane
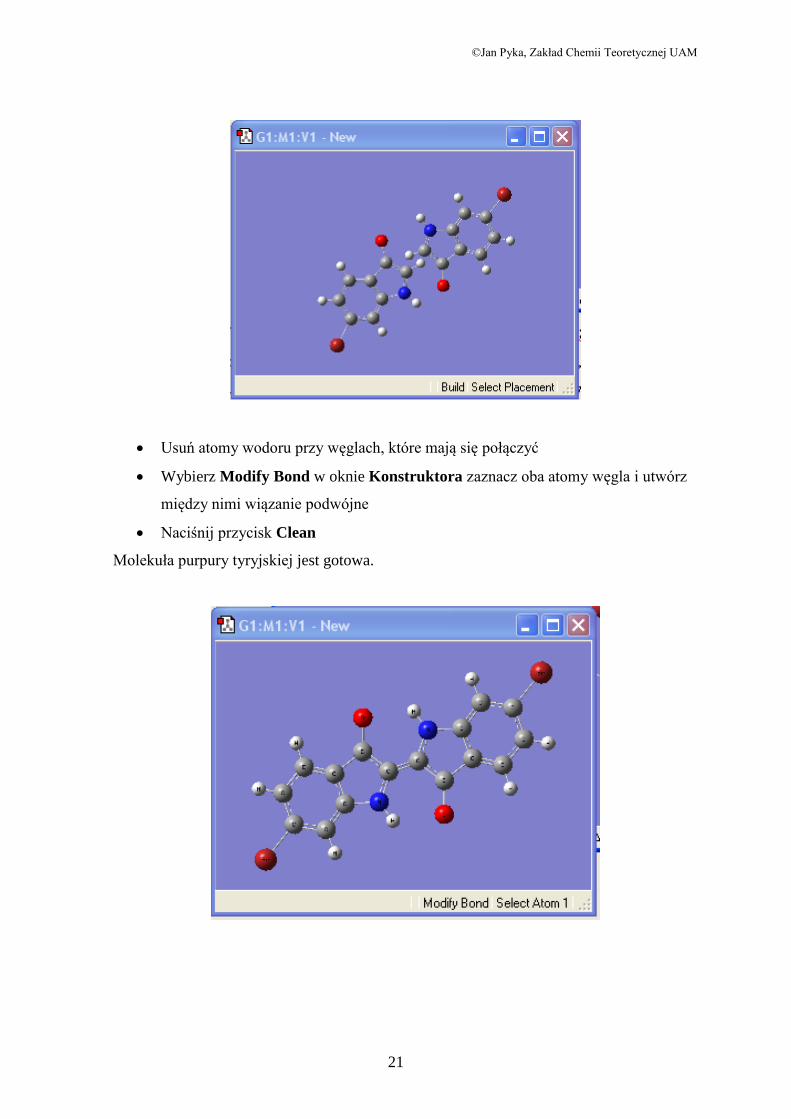
©Jan Pyka, Zakład Chemii Teoretycznej UAM
21
Usuń atomy wodoru przy węglach, które mają się połączyć
Wybierz Modify Bond w oknie Konstruktora zaznacz oba atomy węgla i utwórz
między nimi wiązanie podwójne
Naciśnij przycisk Clean
Molekuła purpury tyryjskiej jest gotowa.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
22
4.4 Łączenie dwóch struktur: budowa dwuchlorodifenylu
Utwórz nowe okno widoku i umieść w nim benzen. Zamień w nim jeden atom
wodoru na atom chloru. Powtórz to samo dla drugiego pierścienia chlorobenzenu.
Obróć drugi pierścień tak, żeby atom chloru znalazł się naprzeciwko tego z
pierścienia pierwszego.
Umieść oba pierścienie w takich pozycjach, że będą się częściowo nakrywały, a
atomy węgla, które mają utworzyć wiązanie znajda się blisko siebie

©Jan Pyka, Zakład Chemii Teoretycznej UAM
23
Usuń atomy wodoru i pozostałe po nich wiązania w obszarze nakrywania
Utwórz wiązanie między węglami, kończąc w te sposób budowę cząsteczki

©Jan Pyka, Zakład Chemii Teoretycznej UAM
24

©Jan Pyka, Zakład Chemii Teoretycznej UAM
25
5 Ustawianie ładunku cząsteczki kationy, aniony Jeśli obliczenia prowadzimy dla kationów, anionów, lub rodników wówczas
przygotowując obliczenia musimy to uwzględnić. Standardowo konstruowana cząsteczka w
programie GaussView posiada sumaryczny ładunek równy zero. Chcąc ustawić inny ładunek
dla cząsteczki z menu Calculate wybieramy opcję Gaussian i w zakładce Method
wpisujemy w opcji Charge ładunek, który chcemy nadać cząsteczce. Musimy także zmienić
multipletowość. Program sam zasugeruje wartość tej zmiennej.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
26
6 Przygotowanie zadania obliczeniowego
6.1 Ustawienia parametrów obliczeń
Po skonstruowaniu cząsteczek w programie GaussView przed zapisem do pliku możemy
ustawić parametry obliczeń. W tym przypadku wybieramy z menu Calculate zakładkę
Gaussian, po czym pojawia się okno:
W oknie tym możemy wybrać rodzaj zadania w zakładce Job Type. Metodę i bazę oraz
dodatkowe parametry ustalamy w zakładce Method. W zakładce Title możemy wpisać
linijkę komentarza, która zostanie umieszczona w plikach z obliczeniami.
Linijki definiujące parametry obliczeń, które ustawiamy, pojawiają się w górnej części
okna:
Title:
©Jan Pyka, Zakład Chemii Teoretycznej UAM
27
Keywords: # hf/3-21g geom=connectivity
Charge/Mult: 0 1
6.2 Wybór rodzaju zadania i metody obliczeń
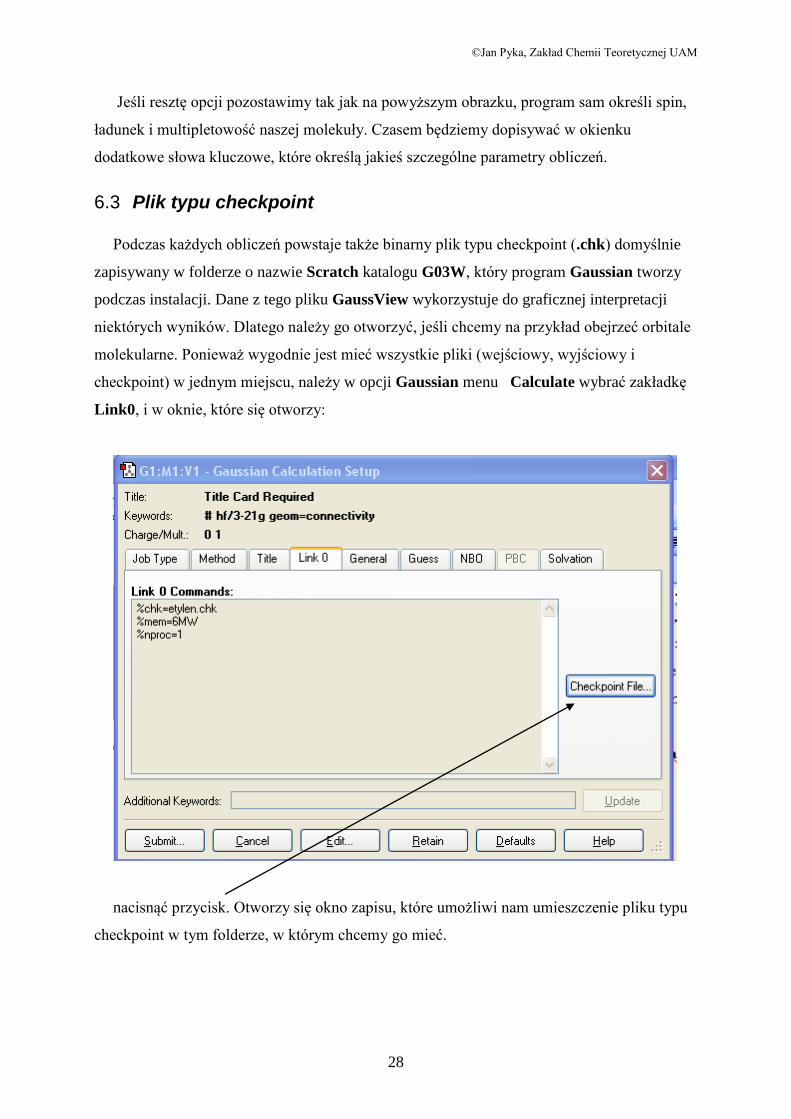
Za pomocą Gaussiana można wykonywa wykonywać wiele typów obliczeń. Wszystkie są
dostępne w rozwijanym menu Job Type. Nas będą interesować zadania:
Energy Obliczanie energii całkowitej (Single Point Energy) stanu
podstawowego molekuły dla jej konkretnej geometrii
Optimization Optymalizacja geometrii cząstek
Frequency Obliczanie częstości drgań molekuł
Frequency + Opt Obliczanie częstości drgań molekuł + optymalizacja geom..
Scan Tworzenie wykresów energii potencjalnej w zależności od
zmian geometrii molekuł.
Oprócz wyboru typu zadania należy jeszcze wybrać metodę obliczeń. W ramach tego
kursu ograniczymy się do obliczeń metodą Hartree-Focka w różnych bazach (Basis Set).
©Jan Pyka, Zakład Chemii Teoretycznej UAM
28
Jeśli resztę opcji pozostawimy tak jak na powyższym obrazku, program sam określi spin,
ładunek i multipletowość naszej molekuły. Czasem będziemy dopisywać w okienku
dodatkowe słowa kluczowe, które określą jakieś szczególne parametry obliczeń.
6.3 Plik typu checkpoint
Podczas każdych obliczeń powstaje także binarny plik typu checkpoint (.chk) domyślnie
zapisywany w folderze o nazwie Scratch katalogu G03W, który program Gaussian tworzy
podczas instalacji. Dane z tego pliku GaussView wykorzystuje do graficznej interpretacji
niektórych wyników. Dlatego należy go otworzyć, jeśli chcemy na przykład obejrzeć orbitale
molekularne. Ponieważ wygodnie jest mieć wszystkie pliki (wejściowy, wyjściowy i
checkpoint) w jednym miejscu, należy w opcji Gaussian menu Calculate wybrać zakładkę
Link0, i w oknie, które się otworzy:
nacisnąć przycisk. Otworzy się okno zapisu, które umożliwi nam umieszczenie pliku typu
checkpoint w tym folderze, w którym chcemy go mieć.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
29
6.4 Zapisywanie pliku do obliczeń
Po skonstruowaniu cząsteczki z zakładki File wybieramy opcję Save i zapisujemy nasz
plik wybierając typ pliku Gaussian Input Files i podajemy nazwę pliku. Dodatkowo
możemy zaznaczyć opcję Write Cartesians. Wówczas struktura cząsteczki zostanie zapisane
w układzie kartezjańskim (przydaje się to do niektórych ćwiczeń).

©Jan Pyka, Zakład Chemii Teoretycznej UAM
30
7 Przykład obliczeń
W ramach przykładu obliczymy teraz energię całkowitą (Single Point Energy) molekuły
formaldehydu (dla wybranej geometrii)
Za pomocą GaussView budujemy molekułę
W menu Calculate wybieramy opcję Gaussian,
Jako Job Type ustalamy Energy
Jako metodę wybieramy Hartree-Fock. Na rysunku poniżej są najbardziej
podstawowe ustawienia parametrów do obliczeń, a przede wszystkim minimalna
baza: 3-21G. O tym, jak należy to rozumieć, dowiecie się na wykładzie, a na razie
pamiętajcie, że im dłuższa baza, tym lepszy wynik (niższą energię) powinniśmy
dostać.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
31
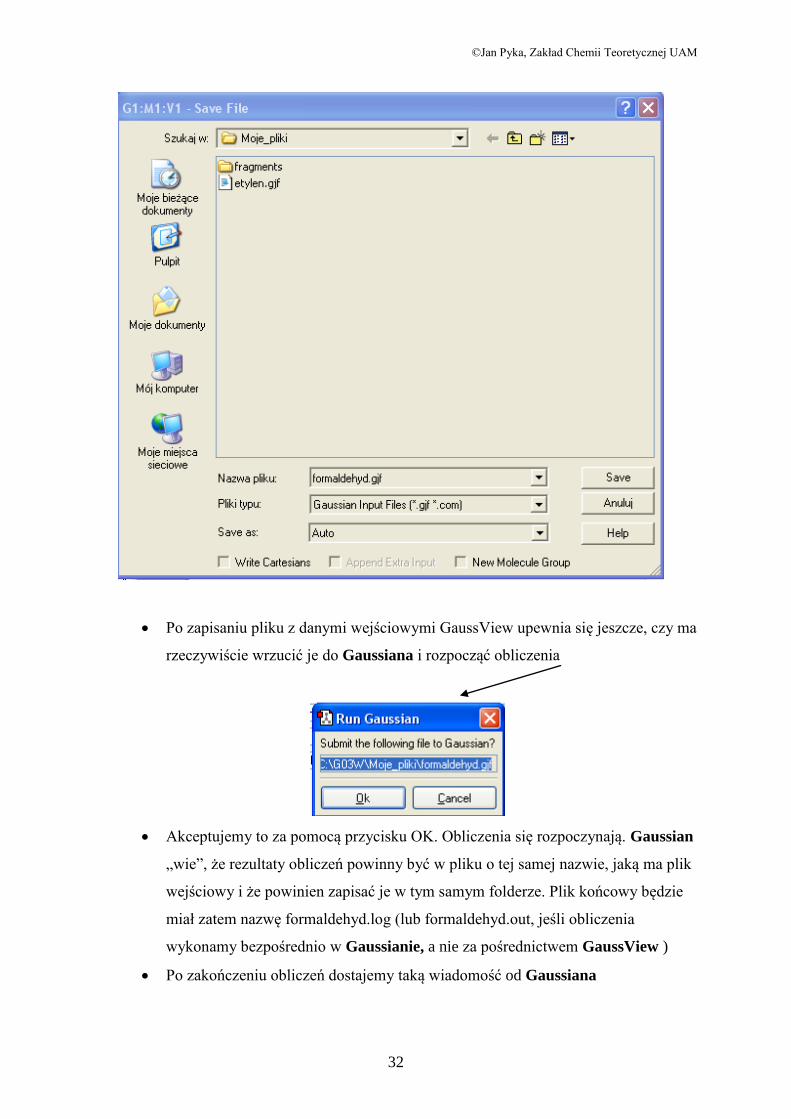
Po przygotowaniu parametrów obliczeń naciskamy przycisk Submit. GaussView
reaguje żądaniem zapisania pliku wejściowego
Naciskamy zatem Save i otwiera się okno zapisu, w którym nadajemy nazwę
naszemu plikowi (formaldehyd.gjf), po czym zapisujemy go w wybranym przez
nas folderze (u mnie jest to folder Moje_pliki)
©Jan Pyka, Zakład Chemii Teoretycznej UAM
32
Po zapisaniu pliku z danymi wejściowymi GaussView upewnia się jeszcze, czy ma
rzeczywiście wrzucić je do Gaussiana i rozpocząć obliczenia
Akceptujemy to za pomocą przycisku OK. Obliczenia się rozpoczynają. Gaussian
„wie”, że rezultaty obliczeń powinny być w pliku o tej samej nazwie, jaką ma plik
wejściowy i że powinien zapisać je w tym samym folderze. Plik końcowy będzie
miał zatem nazwę formaldehyd.log (lub formaldehyd.out, jeśli obliczenia
wykonamy bezpośrednio w Gaussianie, a nie za pośrednictwem GaussView )
Po zakończeniu obliczeń dostajemy taką wiadomość od Gaussiana
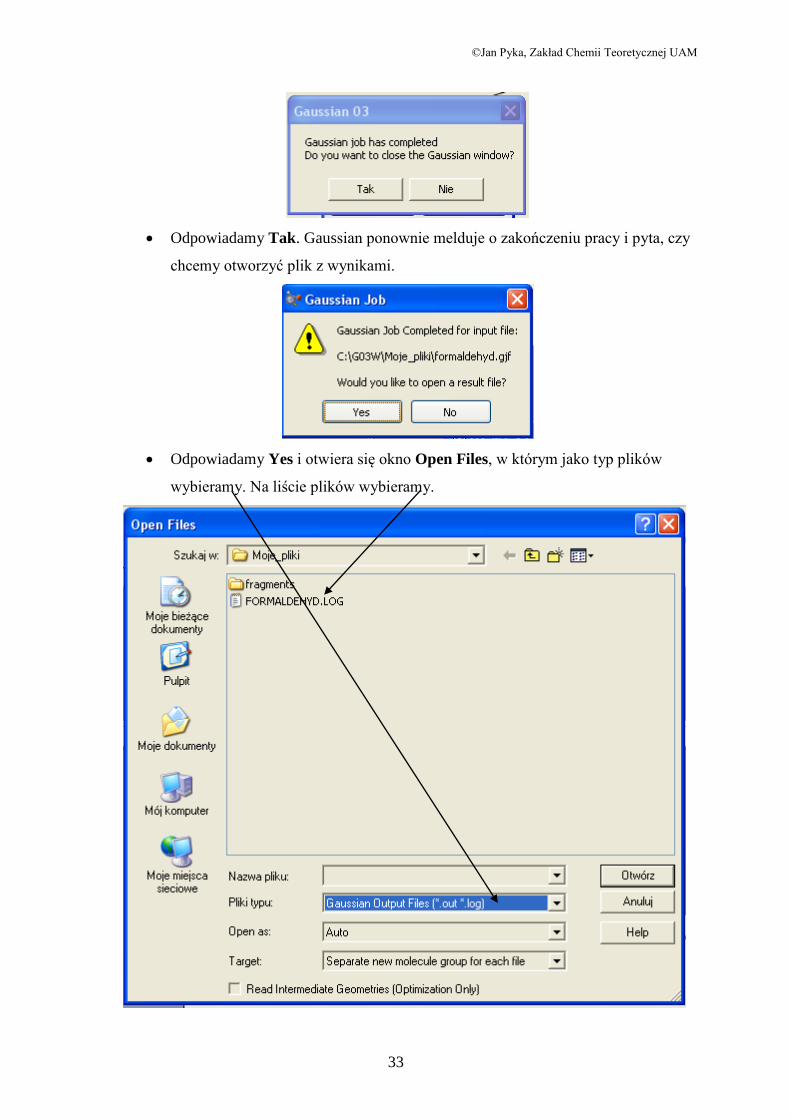
©Jan Pyka, Zakład Chemii Teoretycznej UAM
33
Odpowiadamy Tak. Gaussian ponownie melduje o zakończeniu pracy i pyta, czy
chcemy otworzyć plik z wynikami.
Odpowiadamy Yes i otwiera się okno Open Files, w którym jako typ plików
wybieramy. Na liście plików wybieramy.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
34
Po otwarciu pliku, otwiera się także nowe okno widoku z molekułą, której
geometria została utworzona na podstawie danych wyjściowych.
Obliczoną przez Gaussiana wartość energii można znaleźć w menu Results po
wybraniu opcji Summary. Mamy tu informację, że obliczona metodą RHF
(Restricted Hartree-Fock) energia jest równa -113,22009 a.u.
Ograniczona (Restricted) metoda Hartree-Focka jest zawsze stosowana do
układów zamkniętopowłokowych i o tym, czy wybrać ją, czy też Metodę
Nieograniczoną (Unrestricted) decyduje program GaussView. Wartość energii
podano tutaj w jednostkach atomowych (hartree). Warto pamiętać, że
1 hartree = 4,35983*10-18
J = 27,21165 eV = 6,2773*102 kcal/mol =
= 2,194746*105 cm
-1
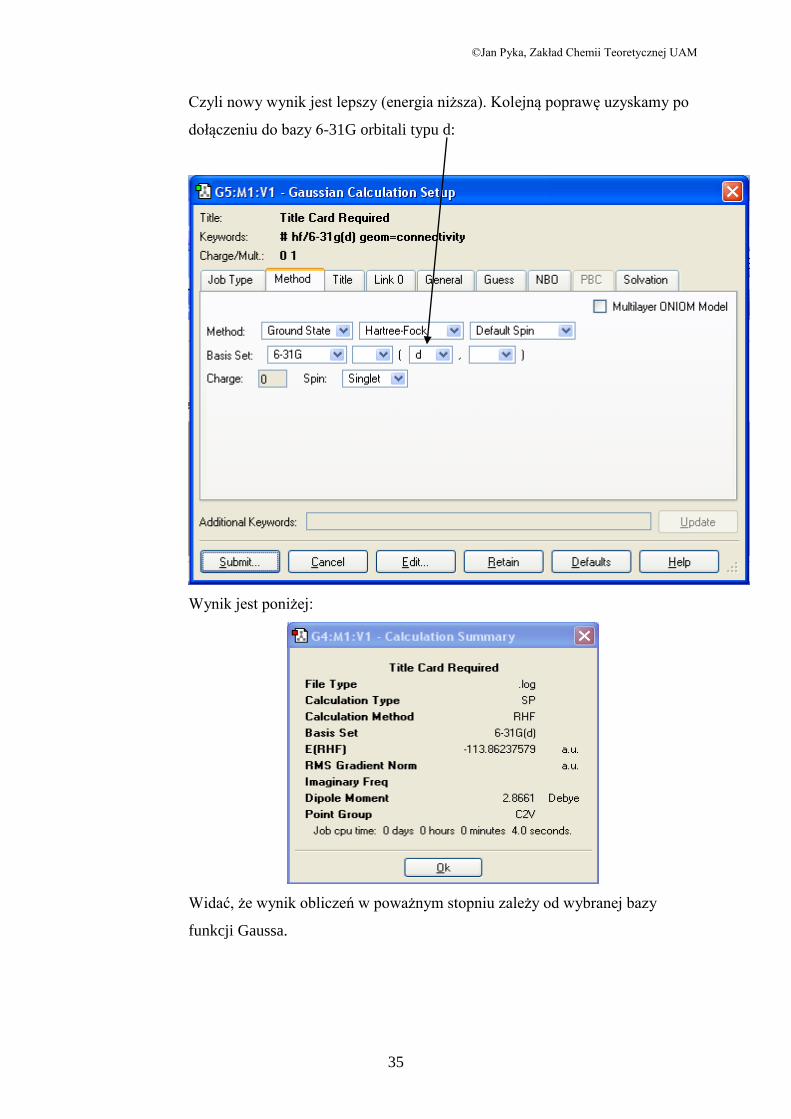
Powtórzmy teraz obliczenia dla bazy 6-31G. Otrzymamy:
©Jan Pyka, Zakład Chemii Teoretycznej UAM
35
Czyli nowy wynik jest lepszy (energia niższa). Kolejną poprawę uzyskamy po
dołączeniu do bazy 6-31G orbitali typu d:
Wynik jest poniżej:
Widać, że wynik obliczeń w poważnym stopniu zależy od wybranej bazy
funkcji Gaussa.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
36
8 Obrazowanie orbitali molekularnych, gęstości elektronowych i potencjałów elektrostatycznych molekuł.
8.1 Orbitale molekularne
W sposób opisany w poprzednich rozdziałach zbudujemy teraz molekułę adeniny i
przeprowadzimy obliczenia energii stanu podstawowego.
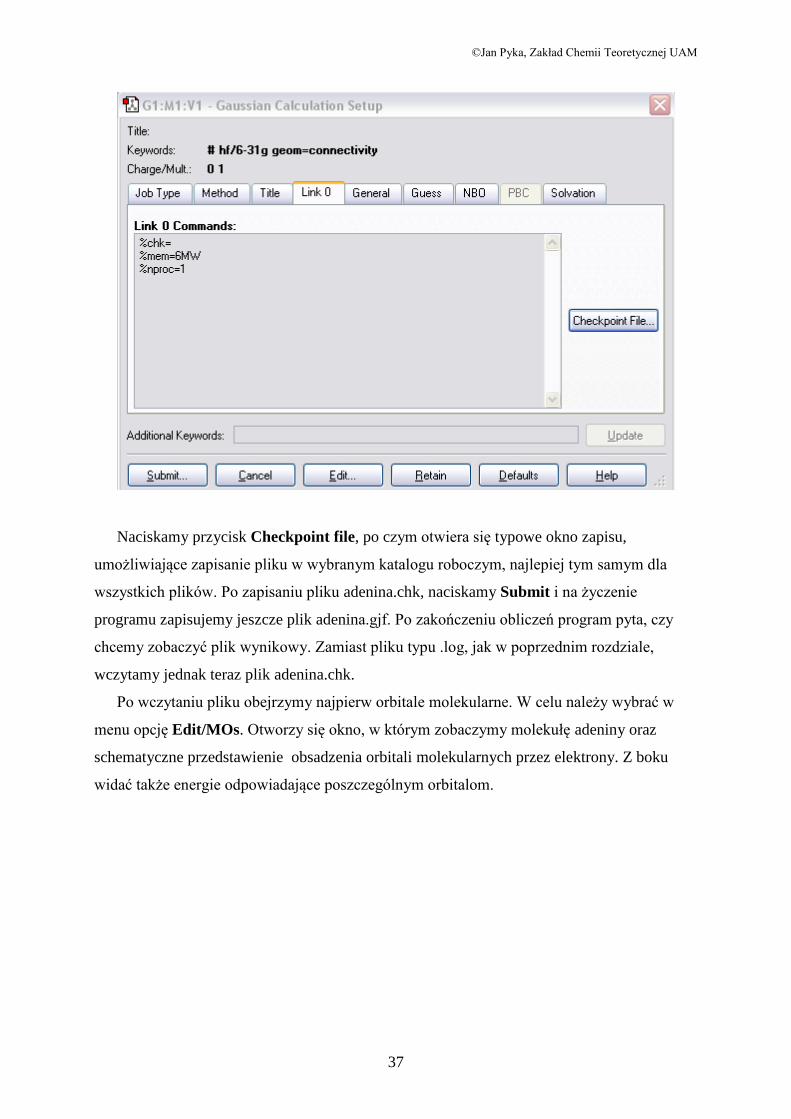
Przed przystąpieniem do obliczeń należy jeszcze wybrać zakładkę Link0,żeby w katalogu
roboczym zapisać plik typu .chk, czyli checkpoint (binarny plik, który zawiera dane
konieczne do stworzenia graficznych przedstawień różnych wielkości charakteryzujących
molekułę). Otworzy się okno:
©Jan Pyka, Zakład Chemii Teoretycznej UAM
37
Naciskamy przycisk Checkpoint file, po czym otwiera się typowe okno zapisu,
umożliwiające zapisanie pliku w wybranym katalogu roboczym, najlepiej tym samym dla
wszystkich plików. Po zapisaniu pliku adenina.chk, naciskamy Submit i na życzenie
programu zapisujemy jeszcze plik adenina.gjf. Po zakończeniu obliczeń program pyta, czy
chcemy zobaczyć plik wynikowy. Zamiast pliku typu .log, jak w poprzednim rozdziale,
wczytamy jednak teraz plik adenina.chk.
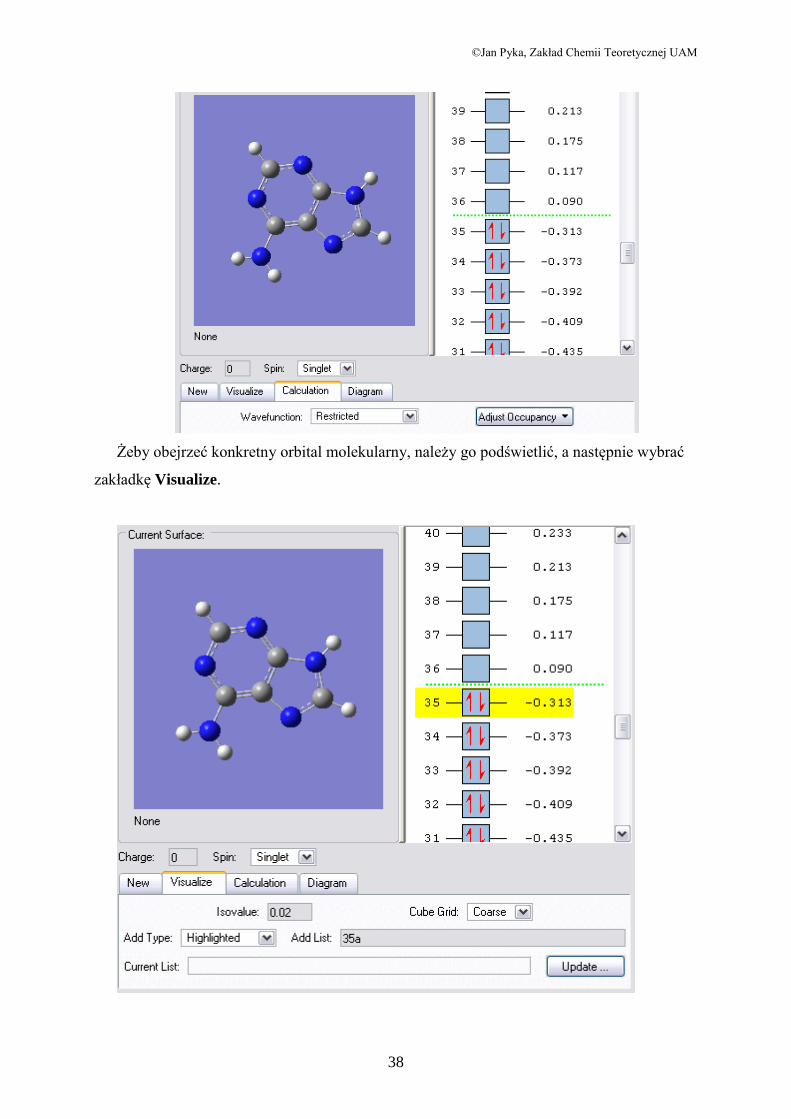
Po wczytaniu pliku obejrzymy najpierw orbitale molekularne. W celu należy wybrać w
menu opcję Edit/MOs. Otworzy się okno, w którym zobaczymy molekułę adeniny oraz
schematyczne przedstawienie obsadzenia orbitali molekularnych przez elektrony. Z boku
widać także energie odpowiadające poszczególnym orbitalom.
©Jan Pyka, Zakład Chemii Teoretycznej UAM
38
Żeby obejrzeć konkretny orbital molekularny, należy go podświetlić, a następnie wybrać
zakładkę Visualize.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
39
W oknie pojawiły się nowe elementy. Po pierwsze dowiadujemy się, że Isovalue jest
równa 0,02. O co tu chodzi? Otóż GaussView przedstawia orbitale molekularne, jak również
inne wielkości (np. gęstość elektronową oraz potencjał elektrostatyczny molekuły) w postaci
warstwic, czyli powierzchni, których punktom odpowiada ta sama, zadana wartość jakiejś
funkcji trzech zmiennych. W tym przypadku przyjęto domyślnie, że ta wartość (izovale) jest
równa 0,02. Druga informacja, która teraz znajdujemy w oknie, to ta, ze Cube Grid jest
Coarse. Co to znaczy. Otóż GaussView oblicza trójwymiarową siatkę wartości danej
wielkości, zwaną Cubes, czyli sześciany, gdyż w narożnikach tych sześcianów program
dokonuje obliczeń. Grid w tym znaczeniu jest informacją o gęstości tej siatki i jeśli
przyjmiemy Coarse, godzimy się na to, że jest dosyć rzadka. Jednak wybranie opcji Medium
lub Fine, wiąże się ze znacznym wydłużeniem czasu obliczeń, a nam tak duża dokładność nie
jest w gruncie rzeczy potrzebna.
Teraz wystarczy nacisnąć na przycisk Update i poczekać, aż program obliczy oraz
wykreśli wybrany przez nas (podświetlony) orbital.
Po naciśnięciu prawego przycisku myszki możemy wybrać opcję Display Format/
Surface i zmienić Solid na np. Transparent.:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
40
W ramach ćwiczeń sprawdźcie teraz, jak wyglądają inne orbitale i jak zmiana Izovalue
wpływa na ich wygląd. Warto także sprawdzić, czy wybór innego Cube Grid w znaczący
sposób powiększa czas obliczeń.
8.2 Gęstość elektronowa
GaussView daje możliwość wykreślenia warstwic całkowitej gęstości elektronowej, czyli,
mówiąc w uproszczeniu, gęstości chmury elektronów w molekule. Chcąc to zrobić, należy
wybrać w menu opcję Results/Surfaces. Otworzy się okno:
Jako Cubes Available (czyli Cubes Dostępne) mamy orbitale molekularny, które
niedawno sobie wykreślaliśmy. Możemy je zachować, albo usunąć, wybierając w Cube
Actions opcję Remove Cube, a potem powtórzyć to samo dla Surface Actions. Jeśli
©Jan Pyka, Zakład Chemii Teoretycznej UAM
41
natomiast zadanie rozpoczniemy od wczytania pliku .chk, nie będzie żadnych dostępnych
wielkości typu cube.
Przystępując do wykreślenia warstwic gęstości elektronowej, musimy najpierw
wyznaczyć trójwymiarowa siatkę jej wartości, czyli w języku GaussView obliczyć
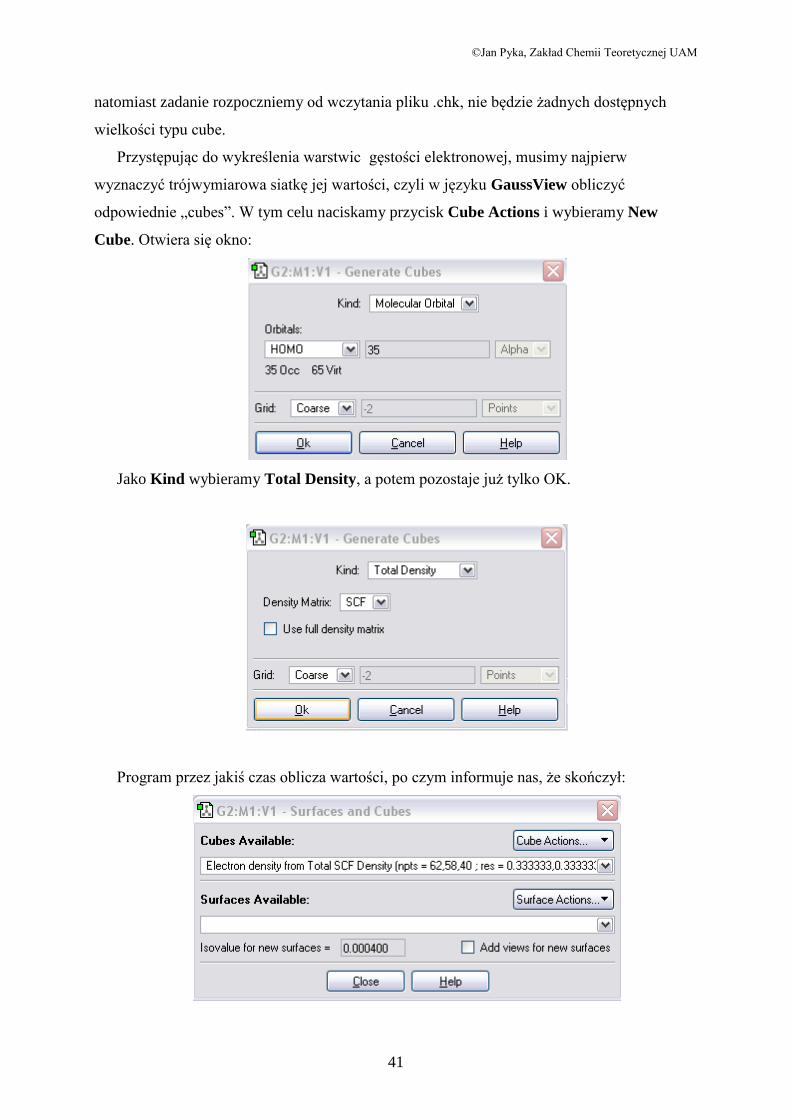
odpowiednie „cubes”. W tym celu naciskamy przycisk Cube Actions i wybieramy New
Cube. Otwiera się okno:
Jako Kind wybieramy Total Density, a potem pozostaje już tylko OK.
Program przez jakiś czas oblicza wartości, po czym informuje nas, że skończył:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
42
Przygotowując się do wykreślenia warstwicy zadajemy odpowiednią Isovalue, warto
także zaznaczyć opcję Add views for new surfaces, dzięki czemu można będzie wykreślić w
różnych oknach kilka wykresów dla np. różnych wartości Isovalue. Następnie w Surface
Actions wybieramy opcję New Surface i program uruchamia obliczenia. Poniżej
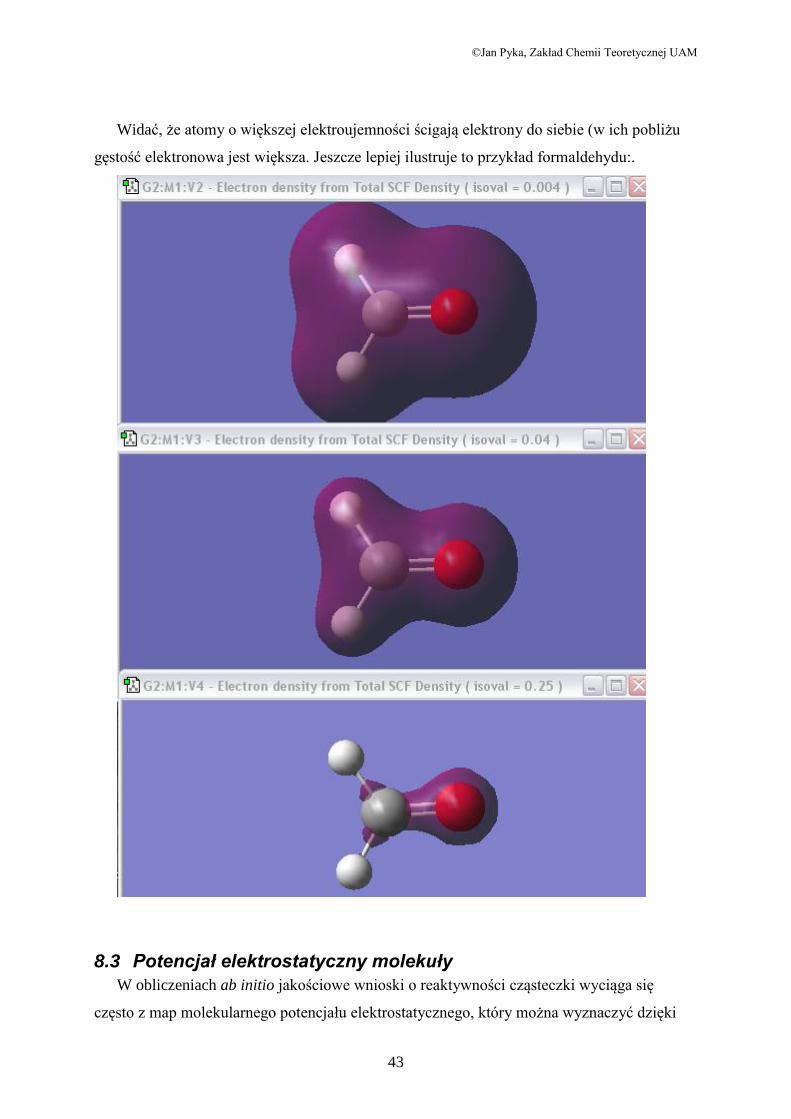
przedstawiam kilka okien dla adeniny oraz (dalej) formaldehydu. W pierwszym wypadku
wybrałem formacie wykresu opcję siatki (mesh), a w drugim transparent.
©Jan Pyka, Zakład Chemii Teoretycznej UAM
43
Widać, że atomy o większej elektroujemności ścigają elektrony do siebie (w ich pobliżu
gęstość elektronowa jest większa. Jeszcze lepiej ilustruje to przykład formaldehydu:.
8.3 Potencjał elektrostatyczny molekuły
W obliczeniach ab initio jakościowe wnioski o reaktywności cząsteczki wyciąga się
często z map molekularnego potencjału elektrostatycznego, który można wyznaczyć dzięki

©Jan Pyka, Zakład Chemii Teoretycznej UAM
44
znajomości gęstości elektronowej. Wykreślimy teraz takie warstwice dla molekuły adeniny.
Po wczytaniu pliku adenina.chk należy wybrać w menu Results/Surfaces, po czym Cube
Actions/New Cube i dalej Kind/ESP
Po naciśnięciu przycisku OK. program oblicza nowe „Cubes” (to może chwilę potrwać), a
po zakończeniu obliczeń zobaczymy infromację:
Teraz można już przygotować warstwicowy wykres potencjału (ESP). W tym celu
zadajemy Isovalue (np. 0,04), po czym naciskamy przycisk Surface Actions i wybieramy
New Surface.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
45
Po chwili otrzymujemy wynik:
Rysunek nasuwa sugestię, że protonowanie adeniny (reakcja elektrofilowa z H+) zachodzi
najłatwiej od strony trzech atomów azotu, przy których potencjał elektrostatyczny ma
największe ujemne wartości (znaki oddają różne kolory warstwic).
GaussView umożliwi także nakładanie (mapping) wartości jednej wielkości na warstwicę
innej, co ułatwia prowadzenie różnych jakościowych rozważań. Dla przykładu nałóżmy
wartości potencjału elektrostatycznego na warstwicę całkowitej gęstości elektronowej
molekuły adeniny. W tym celu należy najpierw obliczyć „Cubes” gęstości elektronowej (jeśli
tego jeszcze nie mamy), a następnie w Surface Actions wybrać New Mapped Surface
©Jan Pyka, Zakład Chemii Teoretycznej UAM
46
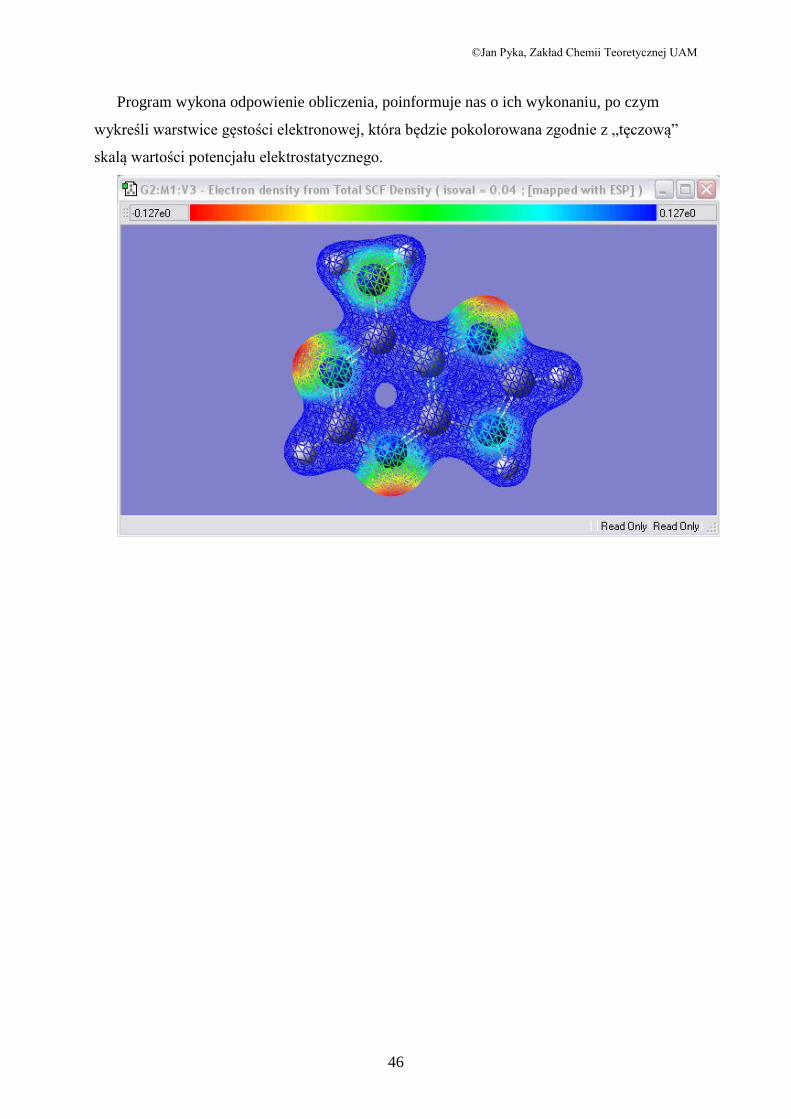
Program wykona odpowienie obliczenia, poinformuje nas o ich wykonaniu, po czym
wykreśli warstwice gęstości elektronowej, która będzie pokolorowana zgodnie z „tęczową”
skalą wartości potencjału elektrostatycznego.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
47
9 Odrobina Teorii
9.1 Metoda Hartree-Focka
W ramach przybliżenia jednoelektronowego najlepsze orbitale (atomowe i molekularne)
uzyskuje się z rozwiązań układu równań Hartree-Focka, które dla układów
zamkniętopowłokowych mają taką postać:
2/,...,2,1)()()( npieiiF ppp
gdzie n to liczba elektronów układu
Przypominają one jednoelektronowe równania Schrodingera, tyle że operator Focka
)()()( iVihiF
, który gra rolę efektywnego hamiltonianu dla i-tego elektronu, jest
operatorem nieco kłopotliwym. Otóż operator )(iV
opisuje uśrednione oddziaływanie i-tego
elektronu z wszystkimi pozostałymi elektronami i chcąc go obliczyć, trzeba znać orbitale
)(ip . Mamy zatem dziwaczną sytuację: do obliczenia któregokolwiek orbitalu p niezbędna
jest znajomość wszystkich orbitali. Wbrew pozorom nie jest to jednak sytuacja beznadziejna.
Problem rozwiązujemy metodą iteracyjną, która umożliwia obliczenie orbitali 2
21 ,..., n z
zadaną dokładnością. Jak to się robi? W metodzie iteracyjnej powtarzamy wielokrotnie
pewien cykl obliczeń i za każdym razem otrzymujemy coraz dokładniejsze rozwiązania, czyli
w tym przypadku orbitale p . Kolejne przybliżenia oznaczymy górnym wskaźnikiem k.
Orbitale k
n
kk
221 ,..., dla
k = 1, 2, 3… wyznaczamy w następujący sposób
1. Zakładamy, kierując się dostępną wiedzą, pewne wyjściowe orbitale, tzn.
k
n
kk
221 ,..., dla k = 0.
2. Stosując powyższe orbitale obliczamy składnik )(iVk
operatora Focka, opisujący
uśrednione oddziaływanie i-tego elektronu z wszystkimi pozostałymi elektronami.
3. Skonstruowany w ten sposób operator Focka (operator
)(ih znamy, gdyż jest to
zwykły hamiltonian jednoelektronowy) wstawiamy do równań Hartree – Focka i

©Jan Pyka, Zakład Chemii Teoretycznej UAM
48
rozwiązujemy je. Otrzymujemy w ten sposób nowy zbiór orbitali w przybliżeniu
k + 1, tj. 1
2
1
2
1
1 ,..., k
n
kk , a także wartości 1k
p .
Po zrealizowaniu punktu 3 wracamy do punktu 2 i stosując otrzymane orbitale obliczamy
potencjał )(iV
w wyższym przybliżeniu, a następnie znowu rozwiązujemy równania Hartree-
Focka, co daje nam nowy zestaw orbitali. Procedurę tę powtarzamy tak długo, aż orbitale
uzyskane przez rozwiązanie równań Hartree-Focka nie różnią się istotnie od orbitali, które
były zastosowane do obliczenia potencjałów występujących w tych równaniach. Dlatego
właśnie metodę Hartree-Focka nazywa się również metodą pola samouzgodnionego
(SCF – Selfconsistent field).
Pozostaje jeszcze odpowiedzieć na pytanie, jak dla założonego potencjału )(iV
rozwiązać
równania Hartree-Focka. Ogólną metodę, która umożliwia przybliżone rozwiązanie tych
równań dla dowolnego potencjału przedstawił Roothan. Zaproponował on, żeby orbitale
Hartree-Focka zapisywać w postaci kombinacji liniowej pewnych znanych funkcji i ,
zwanych bazą:
m
i
ipip c
Podstawiając te rozwinięcia do równań Hartree-Focka otrzymuje się algebraiczne
równania na współczynniki pic . Równania HF z równań różniczkowo-całkowych stają się
równaniami algebraicznymi, których rozwiązanie jest znacznie prostsze.
I tak doszliśmy znowu do Gaussiana, w którym właśnie wykorzystuje się tę metodę, czyli
SCF LCAO MO (Orbitale Molekularne w postaci Liniowych Kombinacji Orbitali
Atomowych uzyskiwane metodą Pola Samouzgodnionego).
9.2 Bazy Orbitali Atomowych
9.2.1 Orbitale slaterowskie (STO –Slater Type Orbitals)
Pierwszym typem orbitali atomowych stosowanych w chemii kwantowej są orbitale
slaterowskie. Nie różnią się one wiele od orbitali atomu wodoru. Pierwszą różnicą jest to, że
w orbitalach slaterowskich część radialna jest znacznie uproszczona. Zamiast wielomianów
Laguerr’a jest po prostu rk , gdzie k jest liczba naturalną lub zerem. Druga i ostatnia różnica

©Jan Pyka, Zakład Chemii Teoretycznej UAM
49
polega na wykładniku orbitalnym , na który nie ma innego ograniczenia poza tym, że ma
być dodatni.
Ogólnie orbitale te mają postać:
)exp(),,()( rzyxfrg
– gdzie f(x,y,z) jest jakimś wielomianem, a parametr orbitalny dobiera się dla
każdego atomu tak, żeby uwzględnić ekranowanie jądra.
Orbitale slaterowskie mają tę wspaniałą zaletę, że zanikają w wraz ze wzrostem r zupełnie
tak samo jak „prawdziwe” orbitale, ale mają też ogromną wadę: całki występujące w
obliczeniach kwantowomechnicznych są niezwykle trudne do policzenie, jeśli używa się tych
orbitali.
9.2.2 Orbitale gaussowskie – GTO (Gaussian Type Orbitals)
Orbitale gaussowskie różnią się od slaterowskich potęgą przy r w wykładniku funkcji
eksponens.:
)exp(),,()( 2rzyxfrg
Mimo tego pozornego utrudnienia dużo łatwiej oblicza się całki kwantowomechaniczne,
kiedy są w nich właśnie orbitale Gaussa.
Najważniejszą przyczyną ogromnego rozwoju chemii kwantowej w ostatnich latach jest to, że
zrezygnowano z funkcji slaterowskich i zwrócono uwagę na orbitale gaussowskie jako
funkcje rozwinięcia.
Jak wskazuje na to sama nazwa, w programie Gaussian używa się funkcji Gaussa. Trzeba
jednak wspomnieć o pewnym ograniczeniu. Otóż orbitale gaussowskie nie zachowują się tak
ładnie jak slaterowskie przy wzroście wartości r – nie zanikają tak samo jak „prawdziwe”
orbitale. Żeby rozwiązać ten problem (oczywiście w przybliżeniu) tworzy się kombinacje
liniowe orbitali gaussowskich, przy czym współczynniki tych rozwinięć są tak dobrane, żeby
kombinacje liniowe zachowywały się w przybliżeniu tak samo, jak orbitale slaterowskie.
Mamy zatem kombinacje liniowe (tzw. orbitale skontraktowane – od angielskiego słowa
contracted, czyli skurczone, zwarte.) funkcji gaussowskich (tzw. prymitywnych gaussianów –
primitive gaussian), które odtwarzają w przybliżeniu kształt orbitali slaterowskich.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
50
W programie Gaussian dostępny jest szeroki zakres baz orbitali skontraktowanych. W
ramach każdej z tych baz każdemu z atomów molekuły przyporządkowywana jest grupa
funkcji, których zadaniem jest przybliżenie „prawdziwych” orbitali. Dla przykładu omówmy
dwie bazy: 6-31G i 6-31(d)
Symbol 6-31G oznacza, że mamy do czynienia z bazą orbitali gaussowskich (G),
kreska pozioma dzieli opis powłok elektronowych w tym wypadku K od L.
Powłoka K jest opisywana pojedynczym orbitalem skontraktowanym, który jest
kombinacją 6 orbitali (prymitywnych) typu 1s, a cyfry 31 dla powłoki L oznaczają
dwa skontraktowane orbitale przypadające na każdy orbital walencyjny (2s, 2px,
2py, 2pz): jeden zawierający trzy GTO i drugi zawierający jeden GTO.
Symbol 6-31(d) oznacza, że w tej bazie użyto dodatkowo orbitali d (tzw. funkcji
polaryzacyjnych).
Funkcje dyfuzyjne (rozmyte) są większą wersją funkcji typu s i p. Pozwalają orbitalom
zajmować większy obszar przestrzeni. Bazy z tymi funkcjami są ważne dla systemów, w
których elektrony są relatywnie daleko od jąder: molekuł z wolnymi parami elektronowymi,
anionów i innych systemów z dużym ładunkiem ujemnym.
Baza 6-31+G(d) to baza 6-31G(d) ze zbiorem funkcji dyfuzyjnych dodanych do ciężkich
atomów. W wersji z podwójnym plusem 6-31++G(d) dodaje się jeszcze funkcje dyfuzyjne do
atomów wodoru.
9.3 Orbitale obsadzone i wirtualne
Ponieważ liczba znalezionych orbitali molekularnych w metodzie SCF LCAO MO jest
równa liczbie użytych orbitali atomowych (skontraktowanych gaussowskich) i , w celu
podwójnego obsadzenia musi być ich co najmniej n/2 (n –liczba elektronów). Zwykle musimy
stosować bazy większe, oprócz więc obsadzonych elektronami orbitali molekularnych
otrzymujemy m – n/2 orbitali nieobsadzonych ( m – liczba funkcji bazowych), które
nazywane są orbitalami wirtualnymi.
Uwaga:
Metoda Hartree-Focka, troszcząc się wyłącznie o energię całkowitą, ignoruje zupełnie
orbitale wirtualne, które w tym rozumieniu są jak gdyby niewykorzystanym produktem
ubocznym metody. Dlatego próba obliczenie energii przejścia elektronowego jako różnicy
energii najniższego orbitalu wirtualnego (LUMO –lowest unoccupied molecular orbital)i

©Jan Pyka, Zakład Chemii Teoretycznej UAM
51
najwyższego obsadzonego orbitalu (HOMO – highest occupied molecular orbital) nie da
dobrego wyniku.
9.4 Ograniczona i nieograniczona metoda Hartree-Focka
Spinorbitale, iloczyny funkcji orbitalnej (orbitala) oraz funkcji spinowej (α dla
magnetycznej liczby spinowej +1/2 lub β dla magnetycznej liczby spinowej -1/2)
φi = ψiα
φi = ψiβ
można konstruować w dwojaki sposób:
zakładając, że część orbitalna dla spinorbitali dwóch sparowanych
elektronów jest taka sama;
nie nakładając takiego warunku.
Pierwszy wariant nazywa się ograniczoną metodą Hartree-Focka (ang. Restricted Hartree-
Fock, RHF), drugi nieograniczoną metodą Hartree-Focka (ang. Unrestricted Hartree-Fock,
UHF). W przypadku obliczeń dla układów zamkniętopowłokowych (stany singletowe)
wykorzystywana jest metoda RHF i otrzymujemy orbitale przestrzenne obsadzone przez pary
sparowanych elektronów. Jeśli z kolei interesują nas układy otwartopowłokowe, dla których
stany podstawowe nie są singletami, GaussView domyślenie (default) wybiera metodę UHF.
W wyniku zastosowania metody UHF otrzymuje się osobno spinorbitale dla elektronów α i
elektronów β. Dla kogoś, kto pierwszy raz zobaczy wyniki, mogą one być trochę zaskakujące,
gdyż na ogół energie orbitalne elektronów tworzących pary są różne. Poniżej przedstawiam
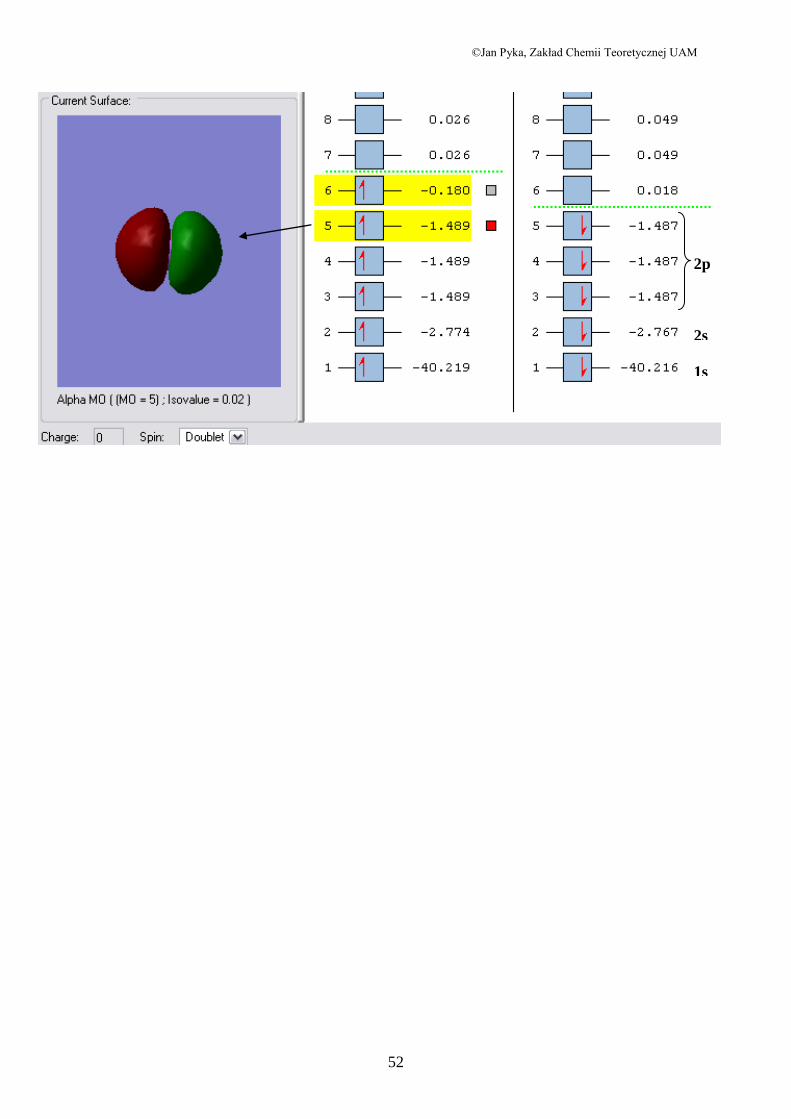
wynik otrzymany dla stanu podstawowego atomu sodu– konfiguracja KL(3s)1
©Jan Pyka, Zakład Chemii Teoretycznej UAM
52
1s
2s
2p

©Jan Pyka, Zakład Chemii Teoretycznej UAM
53
10 Ćwiczenia
10.1 Energia stanu podstawowego atomu wodoru
Chociaż przez cały czas mówimy o orbitalach molekularnych Gaussianem można też bez
kłopotu obliczać energie i orbitale atomów. Atom wodoru jest jedynym rzeczywistym
układem, dla którego można ściśle rozwiązać równanie Schrodingera. Wiemy, że energia
stanu podstawowego atomu wodoru E = -0,5 hartree. Sprawdźmy zatem, jak sprawuje się
Gaussian, który, przypominam, aproksymuje orbitale atomowe funkcjami Gaussa, a zatem
nie możemy oczekiwać dokładnego wyniku.
Zadanie: Przeprowadzić obliczenia energii atomu wodoru metodą HF dla baz: 3-21G,
6-31G, 6-311G.
Wynik:
3-21G -0,4962
6-31G -0,4982
6-311G -0,4998
Widać, że im większa baza, tym lepszy wynik, a dla ostatniej jest niemal doskonały.
GaussView umożliwia także sprawdzenie energii poszczególnych orbitali . W tym celu w
menu Edit należy wybrać opcję MOS. Zobaczymy coś takiego:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
54
Z lewej strony mamy atom, a po środku dość dziwny wykres poziomów molekularnych.
Jest to konsekwencją tego, że atom wodoru ma tylko jeden elektron (multipletowość = 2),
powłoka jest zatem nie zapełniona. W takiej sytuacji GaussView wybiera Nieograniczoną
Metodę Hartree-Focka (Unrestricted Hartree-Fock). W metodzie tej , jak już napisałem w
poprzednim rozdziale, zakłada się, że części przestrzenne spinorbitali elektronów alfa i beta
są różne, co prowadzi do dwóch rozwinięć na funkcje bazowe. W rezultacie trzymuje się dwa
zbiory orbitali molekularnych.
W przypadku atomu wodoru obsadzony jest tylko najniższy orbital alfa o energii równej
energii całkowitej atomu.
Można jeszcze obejrzeć sobie orbital
10.2 Energia stanu podstawowego i orbitale atomowe atomu węgla.
Powtórzymy teraz obliczenia z poprzedniego zadania dla atomu węgla. W obliczeniach
zastosujmy bazę pośrednią 6-31G.
Jeżeli pozwolimy zadecydować programowi GaussView, ustawi on takie parametry
obliczeń:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
55
Jak widać program ustawił multipletowość =1, czyli że w ten sposób obliczymy energię
termu 1S. Ponieważ mamy singlet Gaussian posłuży się metoda RHF (restricted HF), dobrą
dla powłok zamkniętych. Zobaczmy, co nam wyjdzie:
Wynik:
E(RHF) = - 37, 5882 hartree.
A jak obsadzone są orbitale?
©Jan Pyka, Zakład Chemii Teoretycznej UAM
56
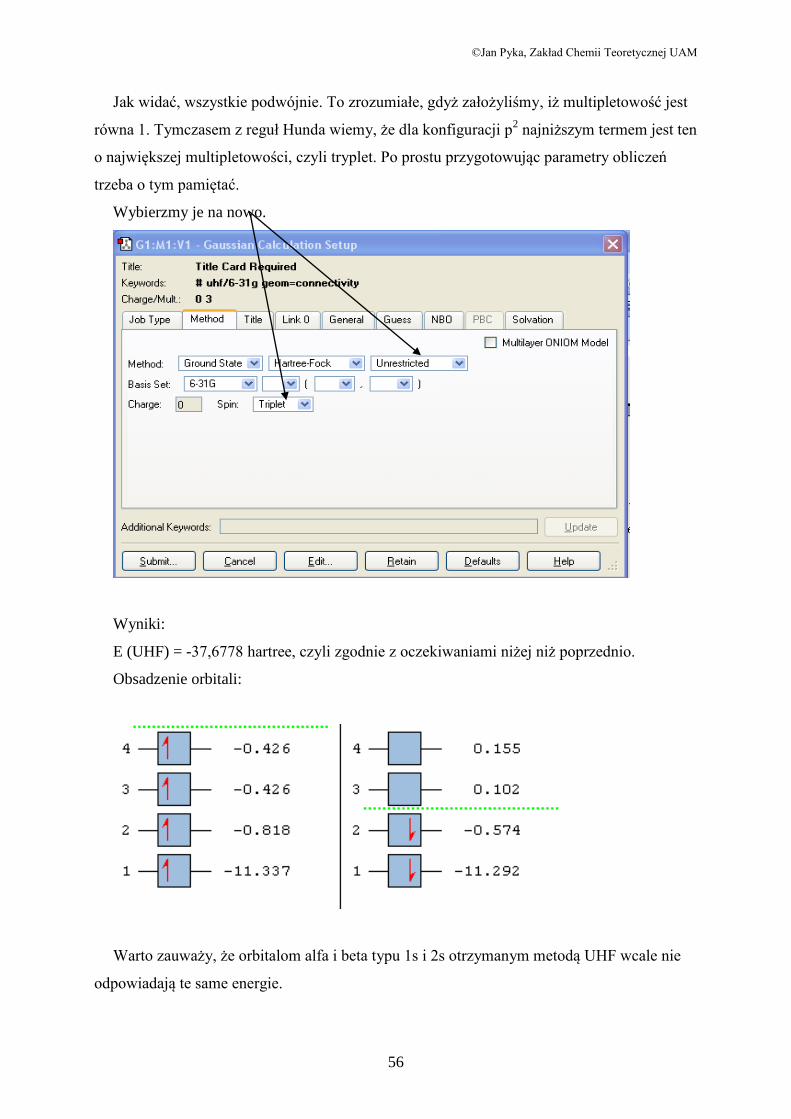
Jak widać, wszystkie podwójnie. To zrozumiałe, gdyż założyliśmy, iż multipletowość jest
równa 1. Tymczasem z reguł Hunda wiemy, że dla konfiguracji p2 najniższym termem jest ten
o największej multipletowości, czyli tryplet. Po prostu przygotowując parametry obliczeń
trzeba o tym pamiętać.
Wybierzmy je na nowo.
Wyniki:
E (UHF) = -37,6778 hartree, czyli zgodnie z oczekiwaniami niżej niż poprzednio.
Obsadzenie orbitali:
Warto zauważy, że orbitalom alfa i beta typu 1s i 2s otrzymanym metodą UHF wcale nie
odpowiadają te same energie.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
57
Na koniec możemy jeszcze obejrzeć orbitale typu 2p, które otrzymaliśmy w wyniku
obliczeń
Oraz
10.3 Konfiguracje elektronowe potasu, wapnia, skandu i tytanu.
Wiadomo, że w rozmieszczeniu energii orbitalnych występują nieregularności i że nawet
ich kolejność może być różna w różnych atomach. Na przykład w podręcznikach chemii
kwantowej można przeczytać, że dla pewnych atomów, np. potasu i wapnia, poziom E4s ma

©Jan Pyka, Zakład Chemii Teoretycznej UAM
58
energię niższą niż poziom E3d. Sprawdźmy to dla atomu potasu, dla którego stan podstawowy
dubletem. Obliczenia przeprowadzimy w bazie 6-31G.
Widać, że podpowłoka 3p jest całkowicie wypełniona, a ostatni elektron zajął orbital 4s,
a nie 3d. Stąd wniosek, że E4s jest rzeczywiście niższa od E3d, a konfiguracją elektronową
potasu w stanie podstawowym jest:
K: (1s)2(2s)
2(2p)
6(3s)
2(3p)
6(4s)
1
Zadanie: Sprawdzić, jak to wygląda dla wapnia. Potwierdzono eksperymentalnie, że stan
podstawowy wapnia jest singletem.
Podpowiedź:
Przeprowadźcie obliczenia dla singletu i trypletu. Porównajcie energie całkowite.
Wizualizując najwyższe obsadzone orbitale, ustalcie, czy są to stany 4s, czy też 3d.
Dla stanu podstawowego powinniście otrzymać konfigurację:
(1s)2(2s)
2(2p)
6(3s)
2(3p)
6(4s)
2
10.4 Energie stanu podstawowego atomów i jonów pierwszego szeregu
Pamiętając o konieczności zmiany ładunku, odtwórz wyniki z poniższej tabeli:
3-21G 6-31G Eexp
He -2,836 -2,855 -2,904
3p
4s

©Jan Pyka, Zakład Chemii Teoretycznej UAM
59
Li+ -7,187 -7,236 -7,280
Be2+
-13,530 -13,610 -13,657
B3+
-21,863 -21,984 -22,035
10.5 Badania molekuł
W wypadku molekuł całkowita energia jest zależna od odległości między jądrami
molekuły (konkretnej geometrii) i dlatego nazywa się ją „single point energy” Jest to
konsekwencją tego, że w przybliżeniu Borna-Oppenheimera (a właściwie adiabatycznym),
elektronowe równanie Schrodingera rozwiązuje się dla ustalonej konfiguracji jąder.
10.5.1 Obliczanie energii dla określonej geometrii cząsteczki (single point energy)
Etylen
Obliczymy teraz energię etylenu dla konkretnej geometrii, którą otrzymamy z
GaussView.
W tablicy rodników znajdujemy winyl
Po skopiowaniu do okna widokowego zmienia się w etylen.
W menu Calculate wybieramy Gaussian, a w zakładce Job Type: Energy
Przechodzimy do zakładki Metod, w której ustalamy parametry obliczeń

©Jan Pyka, Zakład Chemii Teoretycznej UAM
60
W zakładce Link0 ustalamy ścieżkę do zapisu pliku checkpoint.(nazwę)
Naciskamy Submit i uruchamiamy obliczenia
Otrzymujemy energię: E = -78.030 hartree i moment dipolowy równy zero. Cząsteczka
zostaje też ustawiona w tzw. standardowej orientacji, co mian sobie obejrzeć po włączeniu osi
kartezjańskich: leży w płaszczyźnie xy, a wiązanie C–C pokrywa się z osią y.
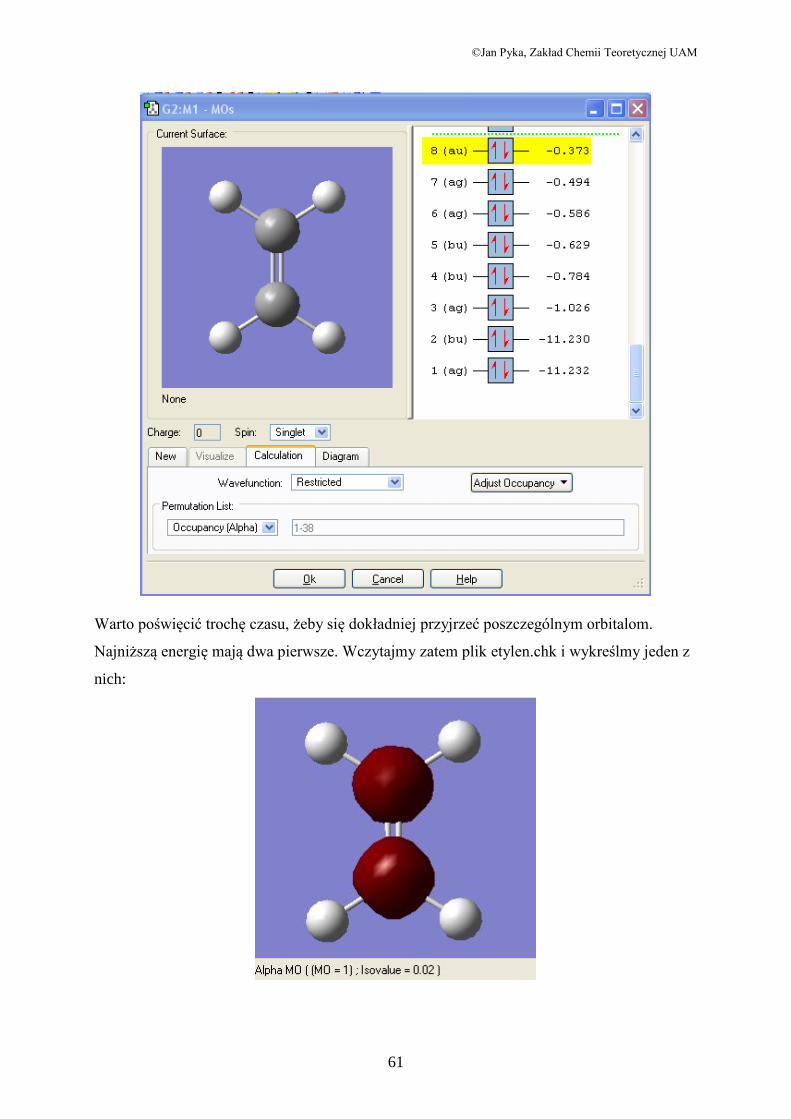
Orbitale molekularne:
W menu Edit znajdujemy opcję MOs, po czym otwiera się okno:
©Jan Pyka, Zakład Chemii Teoretycznej UAM
61
Warto poświęcić trochę czasu, żeby się dokładniej przyjrzeć poszczególnym orbitalom.
Najniższą energię mają dwa pierwsze. Wczytajmy zatem plik etylen.chk i wykreślmy jeden z
nich:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
62
Zgodnie z oczekiwaniami widać, że składa się z dwóch orbitali atomowych,
scentrowanych na atomach węgla i nie biorących żadnego udziału w tworzeniu wiązań
molekularnych.
Dla porównania ósmy, najwyższy orbital molekularny (HOMO)
Widać, że jest to orbital obejmujący cała molekułę, utworzony głównie z orbitali pz obu
atomów węgla. W HOMO orbitale atomowe wchodzą do kombinacji liniowej z tymi samymi
znakami i tworzą wiążący orbital typu . W LUMO (najniższy nie obsadzony orbital
molekularny) mają one przeciwne znaki i tworzą antywiążacy orbital , który wygląda tak:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
63
Formaldehyd
Dla porównania powtórzymy te obliczenia dla formaldehydu
Cząsteczkę zbudujemy, wykorzystując tablicę rodników
Obliczenia przeprowadzamy tak samo, jak to zrobiliśmy dla etylenu
W wyniku obliczeń otrzymujemy energię.
E = - 113,857 hartree
Molekuła została także ustawiano standardowej orientacji: cząsteczka leży na płaszczyźnie
yz, przy czym wiązanie C–O pokrywa się z osią z.
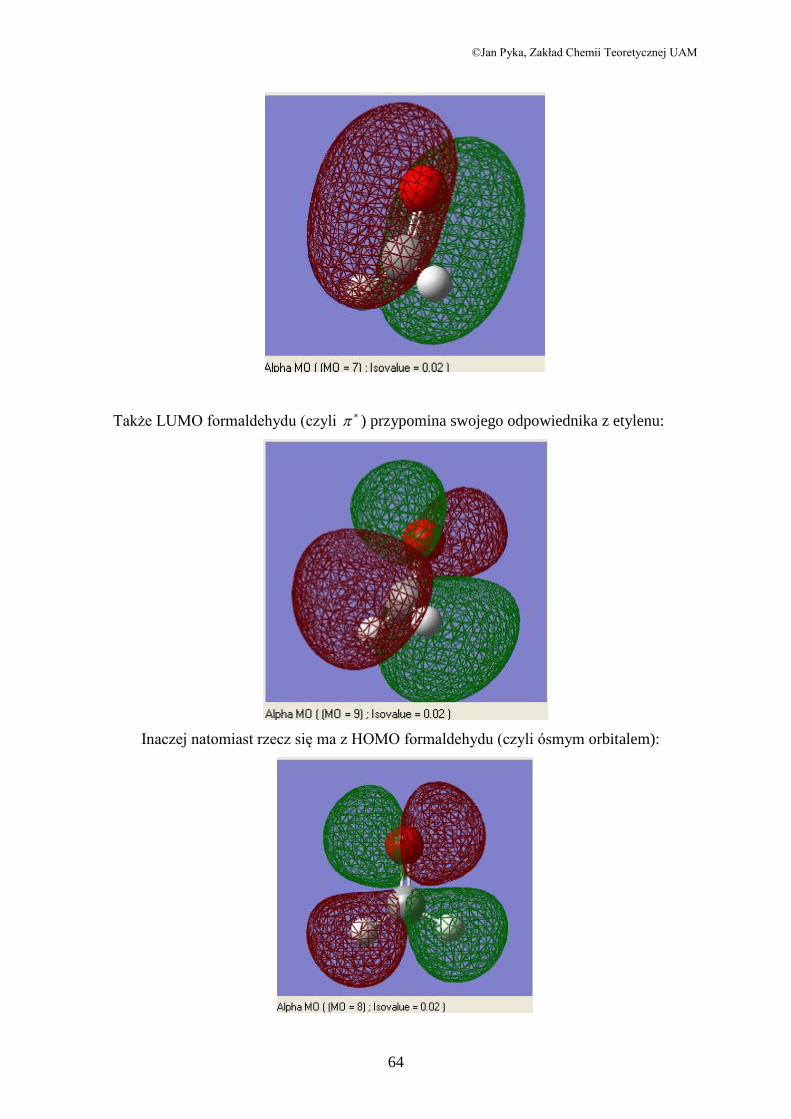
A teraz orbitale molekularne. Orbital siódmy formaldehydu ma te same cechy co orbital
HOMO etylenu i jest także wiążącym orbitalem . Widać go poniżej. Przedstawiłem go
trochę inaczej niż orbital etylenu; zamiast ciągłej powierzchni (solid) zastosowałem w
formacie siatkę (mesh), żeby lepiej było widać molekułę.
©Jan Pyka, Zakład Chemii Teoretycznej UAM
64
Także LUMO formaldehydu (czyli ) przypomina swojego odpowiednika z etylenu:
Inaczej natomiast rzecz się ma z HOMO formaldehydu (czyli ósmym orbitalem):

©Jan Pyka, Zakład Chemii Teoretycznej UAM
65
Jest to orbital utworzony z orbitali atomowych py węgla i tlenu oraz orbitali typu s atomów
wodoru. Widać, że w przeciwieństwie do HOMO etylenu nie jest on antysymetryczny
względem odbicia w płaszczyźnie cząsteczki, czyli nie jest orbitalem . Różnica wynika z
obecności dwóch wolnych par elektronowych atomu tlenu. Z sześciu elektronów
walencyjnych tego atomu, dwa biorą udział w tworzeniu podwójnego wiązania z węglem, a
cztery istnieją jako dwie wolne pary.
10.6 „Ręczna” Optymalizacja długości wiązań dla cząsteczki wody
Cząsteczka wody ma dwa geometryczne parametry, których wartość należy określić, żeby
sprecyzować jej strukturę: długości wiązań O-H i wielkość kąta H–O–H. W tym ćwiczeniu
wykonamy szereg obliczeń, żeby zobaczyć, jak wartość energii całkowitej (w stanie
podstawowym molekuły) zależy od długości wiązań O-H. Wielkość kąta pozostanie stała i
równa 109 stopni. Długości wiązań będziemy zmieniać od 0,8 angstrema do 1,5 angstema z
krokiem 0,05. Obliczone wartości energii należy zapisywać. Po zakończeniu obliczeń
sporządzimy wykres energii w Excelu..
W pierwszym kroku budujemy molekułę wody i wprowadzamy zadane wartości
parametrów geometrycznych.
Przechodzimy do menu Calculate programu GaussView, wybieramy opcję
Gaussian, a w zakładce Job Type: Energy
W zakładce Method ustalamy następujące parametry obliczeń:
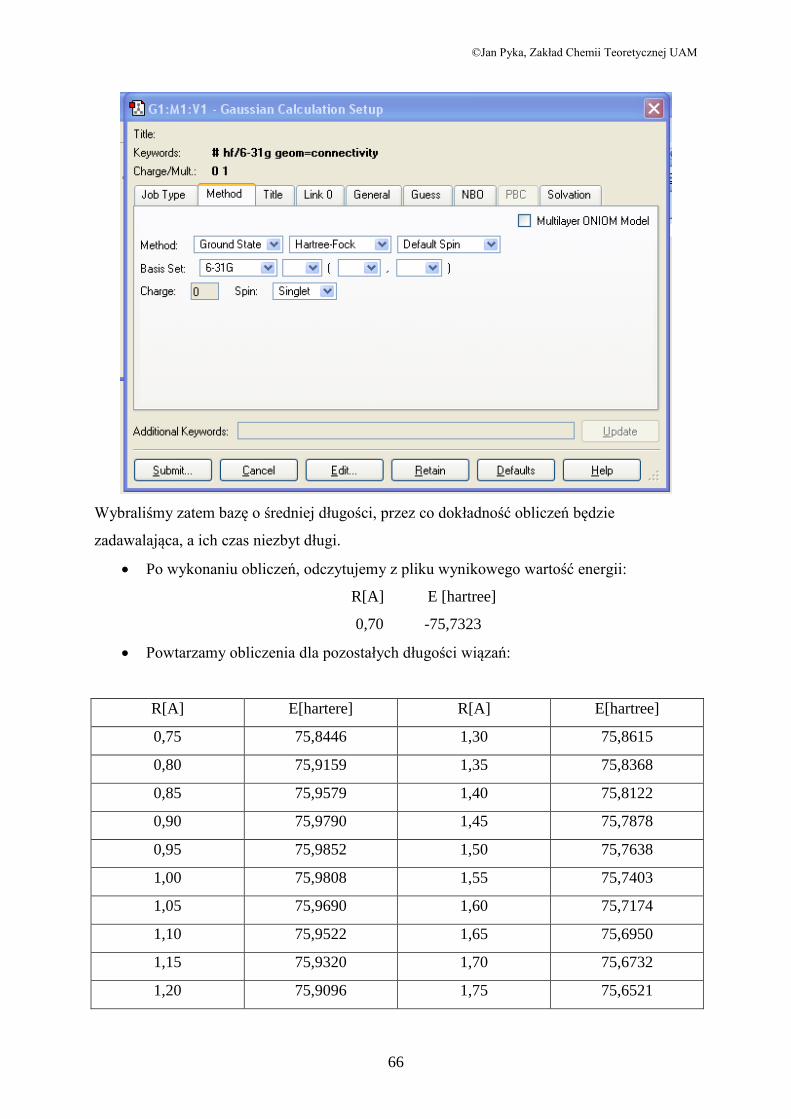
©Jan Pyka, Zakład Chemii Teoretycznej UAM
66
Wybraliśmy zatem bazę o średniej długości, przez co dokładność obliczeń będzie
zadawalająca, a ich czas niezbyt długi.
Po wykonaniu obliczeń, odczytujemy z pliku wynikowego wartość energii:
R[A] E [hartree]
0,70 -75,7323
Powtarzamy obliczenia dla pozostałych długości wiązań:
R[A] E[hartere] R[A] E[hartree]
0,75 75,8446 1,30 75,8615
0,80 75,9159 1,35 75,8368
0,85 75,9579 1,40 75,8122
0,90 75,9790 1,45 75,7878
0,95 75,9852 1,50 75,7638
1,00 75,9808 1,55 75,7403
1,05 75,9690 1,60 75,7174
1,10 75,9522 1,65 75,6950
1,15 75,9320 1,70 75,6732
1,20 75,9096 1,75 75,6521

©Jan Pyka, Zakład Chemii Teoretycznej UAM
67
1,25 75,8859
Jeżeli wykreślimy te dane w Excelu, otrzymamy znany kształt krzywej potencjału – należy
pamiętać, że energia elektronowa gra rolę potencjału dla ruchu jąder.
-76,05
-76,00
-75,95
-75,90
-75,85
-75,80
-75,75
-75,70
-75,65
-75,60
0,00 0,50 1,00 1,50 2,00
Serie1
Z powyższego zadania wynika, że dla ustalonego kąta optymalną (równowagową)
długością obu wiązań jest wartość zbliżona do 0,9 A.
10.7 Wykres (scan) energii potencjalnej
Zadanie, które wykonaliśmy w poprzednim podrozdziale można oczywiście
zautomatyzować, żeby nie było konieczności wielokrotnego powtarzania tych samych
obliczeń. Zróbmy to teraz na przykładzie molekuły H2, postępując zgodnie z poniższymi
instrukcjami:
Budujmy molekułę:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
68
Zmieniamy długość wiązania na 0,4 Angstrema
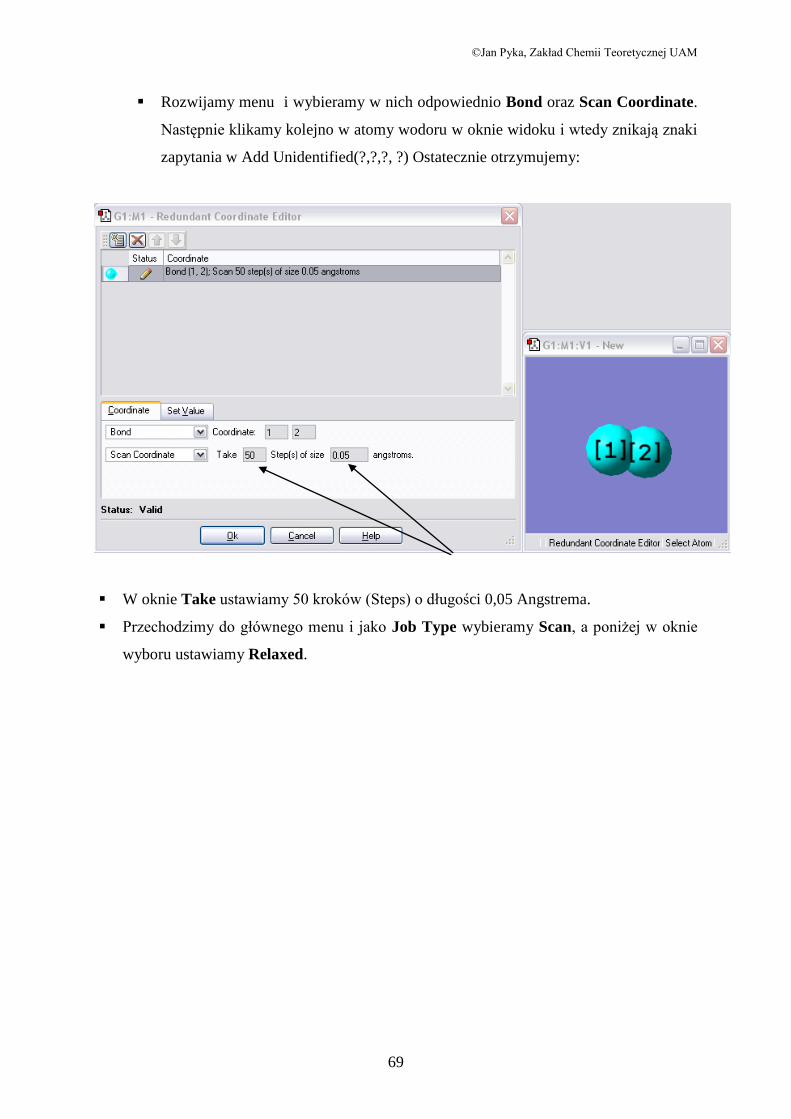
W Menu Edit otwieramy okno Redundant Coordinate Editor
Po naciśnięciu na wskazaną przez strzałkę ikonę, okno przybiera postać
©Jan Pyka, Zakład Chemii Teoretycznej UAM
69
Rozwijamy menu i wybieramy w nich odpowiednio Bond oraz Scan Coordinate.
Następnie klikamy kolejno w atomy wodoru w oknie widoku i wtedy znikają znaki
zapytania w Add Unidentified(?,?,?, ?) Ostatecznie otrzymujemy:
W oknie Take ustawiamy 50 kroków (Steps) o długości 0,05 Angstrema.
Przechodzimy do głównego menu i jako Job Type wybieramy Scan, a poniżej w oknie
wyboru ustawiamy Relaxed.
©Jan Pyka, Zakład Chemii Teoretycznej UAM
70
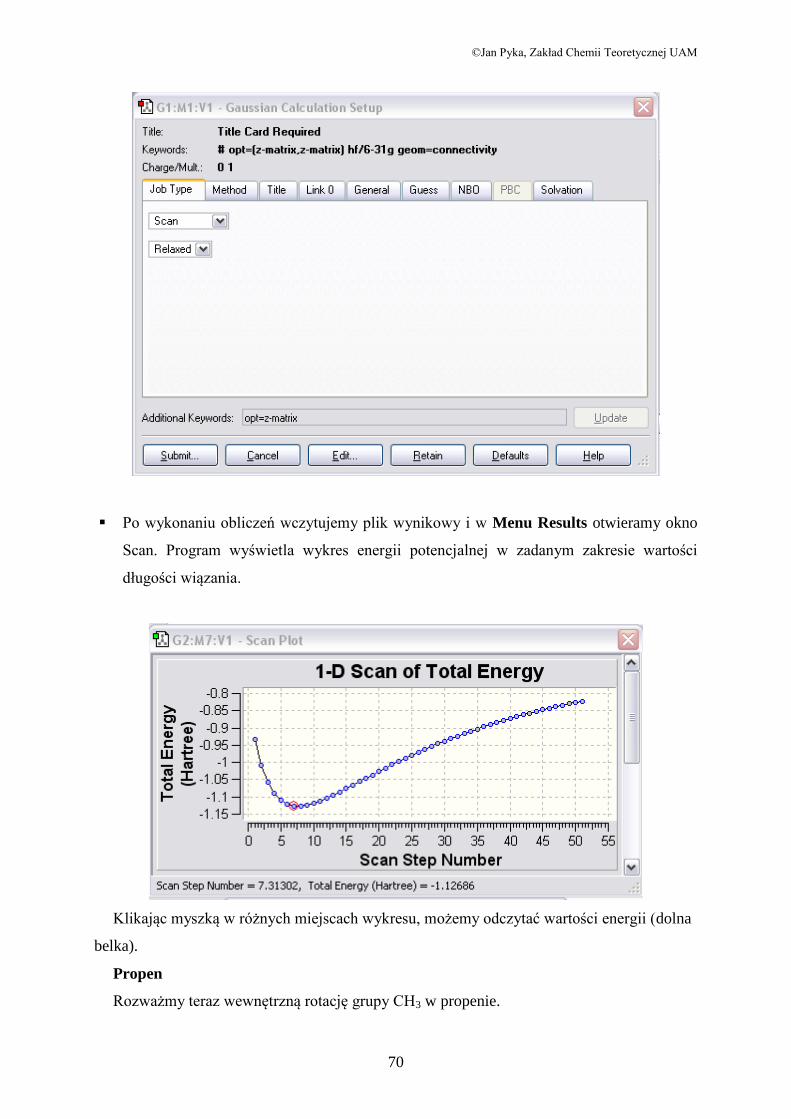
Po wykonaniu obliczeń wczytujemy plik wynikowy i w Menu Results otwieramy okno
Scan. Program wyświetla wykres energii potencjalnej w zadanym zakresie wartości
długości wiązania.
Klikając myszką w różnych miejscach wykresu, możemy odczytać wartości energii (dolna
belka).
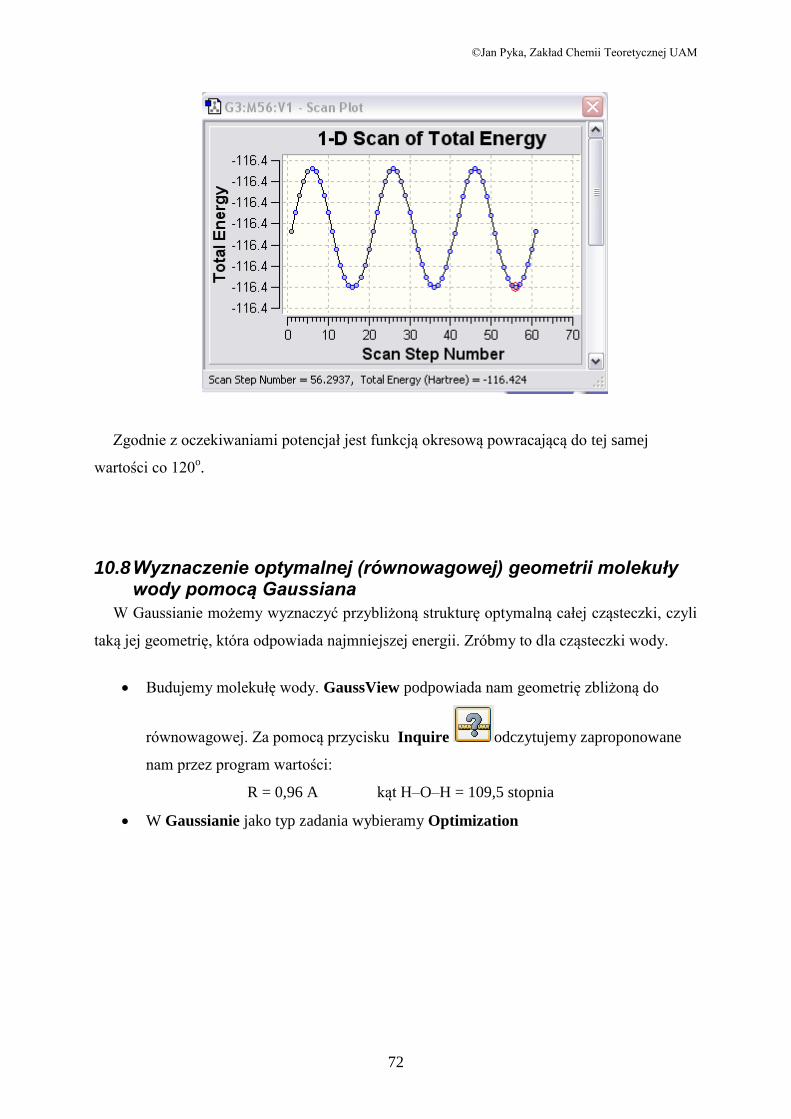
Propen
Rozważmy teraz wewnętrzną rotację grupy CH3 w propenie.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
71
Zmienną jest kąt dwuścienny zdefiniowany przez atomy 1,2,3 4. Ustalamy, że chcemy
zbadać wykres energii potencjalnej w zakresie 360o poczynając od -90
o, z krokiem 6
o.
Po kilkunastominutowych obliczeniach otrzymujemy:
©Jan Pyka, Zakład Chemii Teoretycznej UAM
72
Zgodnie z oczekiwaniami potencjał jest funkcją okresową powracającą do tej samej
wartości co 120o.
10.8 Wyznaczenie optymalnej (równowagowej) geometrii molekuły wody pomocą Gaussiana
W Gaussianie możemy wyznaczyć przybliżoną strukturę optymalną całej cząsteczki, czyli
taką jej geometrię, która odpowiada najmniejszej energii. Zróbmy to dla cząsteczki wody.
Budujemy molekułę wody. GaussView podpowiada nam geometrię zbliżoną do
równowagowej. Za pomocą przycisku Inquire odczytujemy zaproponowane
nam przez program wartości:
R = 0,96 A kąt H–O–H = 109,5 stopnia
W Gaussianie jako typ zadania wybieramy Optimization

©Jan Pyka, Zakład Chemii Teoretycznej UAM
73
a w Method znowu wybieramy obliczenia metodą HF dla stanu podstawowego w bazie
6-31G.
Program znajduje minimum energii w punkcie E = -75,9854 dla długości wiązań i kąta (w
zaokrągleniu):
R = 0,95 A Kąt H–O–H = 111,54 stopnia
Jeśli porówna to z wielością kąta wyznaczoną eksperymentalnie, czyli 104,5o, wynik może
się wydawać niezbyt satysfakcjonujący, ale należy pamiętać, że uzyskaliśmy to za pomocą
niezbyt długiej bazy. Rozbudujmy ją nieco, dodając orbitale d i tzw. funkcje rozmyte
(dyfuzyjne) . Uzyskamy to przez podwójny znaczek ++, który dodaje funkcje dyfuzyjne do
tlenu (pierwszy +) i obu wodorów (drugi +).

©Jan Pyka, Zakład Chemii Teoretycznej UAM
74
Po wykonaniu obliczeń, otrzymujemy energię minimalną :
E = -76,0179
czyli niższą, a zatem lepszą niż poprzednio. Geometria też jest o wiele bliższa
eksperymentalnej: R= 0.948 A kąt H–O–H = 106,465
Zadanie 1: Wyznaczyć geometrię równowagową molekuły etylenu.
Rozwiązanie:
Budujemy molekułę i wykorzystując prawoklik włączamy opcję wyświetlającą Labels
(numery atomów)

©Jan Pyka, Zakład Chemii Teoretycznej UAM
75
Wykorzystując przycisk Inquire możemy teraz odczytać kilka
początkowych wartości. I tak :
R(1,2) - 1,326 Ā
R(1,4) - 1,098 Ā
A(3,1,2) - 122,716o
Zastosujmy jeszcze opcję Clean, a potem Symmetrize (symetryzacja). Teraz
odpowiednie parametry wejściowe mają inne wartości, np.:
R(1,2) - 1,352 Ā
R(1,4) - 1,070 Ā
A(3,1,2) - 120.0o
Wykonujemy obliczenia i po przeprowadzeniu optymalizacji w bazie 6-31G(d)
otrzymujemy następującą geometrię:
R(1,2) - 1,317 Ā
R(1,4) - 1,076 Ā
A(3,1,2) - 121,8o
A(4,1,2) - 121,8o
A(3,1,4) - 116,4o
Kolejno odczytujemy także inne wartości.
Zadanie 2. Znaleźć geometrię równowagową fluoroetylenu.
Rozwiązanie. Budujemy cząsteczkę i zamieniamy jeden z wodorów na fluor. Po włączeniu
numeracji otrzymujemy następująca strukturę. Atom o numerze 6 to oczywiście fluor.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
76
Po wykonaniu obliczeń (tym razem nie symetryzowałem cząsteczki) powinniście
otrzymać:
R(1,2) - 1,31 Ā
R(1,4) - 1,33 Ā
A(6,1,2) - 122,4o
A(3,1,2) - 125,7
A(3,1,6) - 111,9o
Porównując wyniki otrzymane dla molekuł etylenu i fluoroetylenu możemy sprawdzić, jak
na ich geometrię wpływa zastąpienie jednego z wodorów etylenem.
©Jan Pyka, Zakład Chemii Teoretycznej UAM
77
10.9 Optymalizacja geometrii cząsteczek II
W poprzednich zadaniach wyznaczyliśmy zależność energii cząsteczek w zależności od
jednej ze współrzędnych (długość wiązań O-H w wodzie i kąt wewnętrznej rotacji w
propenie) i w obu wypadkach otrzymaliśmy wykres funkcji jednej zmiennej, zwany
zwyczajowo „powierzchnią energii potencjalnej”. Powierzchnia energii potencjalnej jest
funkcją opisującą zależność energii molekuły od jej geometrii i dla większych systemów jest
zależna od tylu zmiennych ile jest stopni swobody w cząsteczkach.
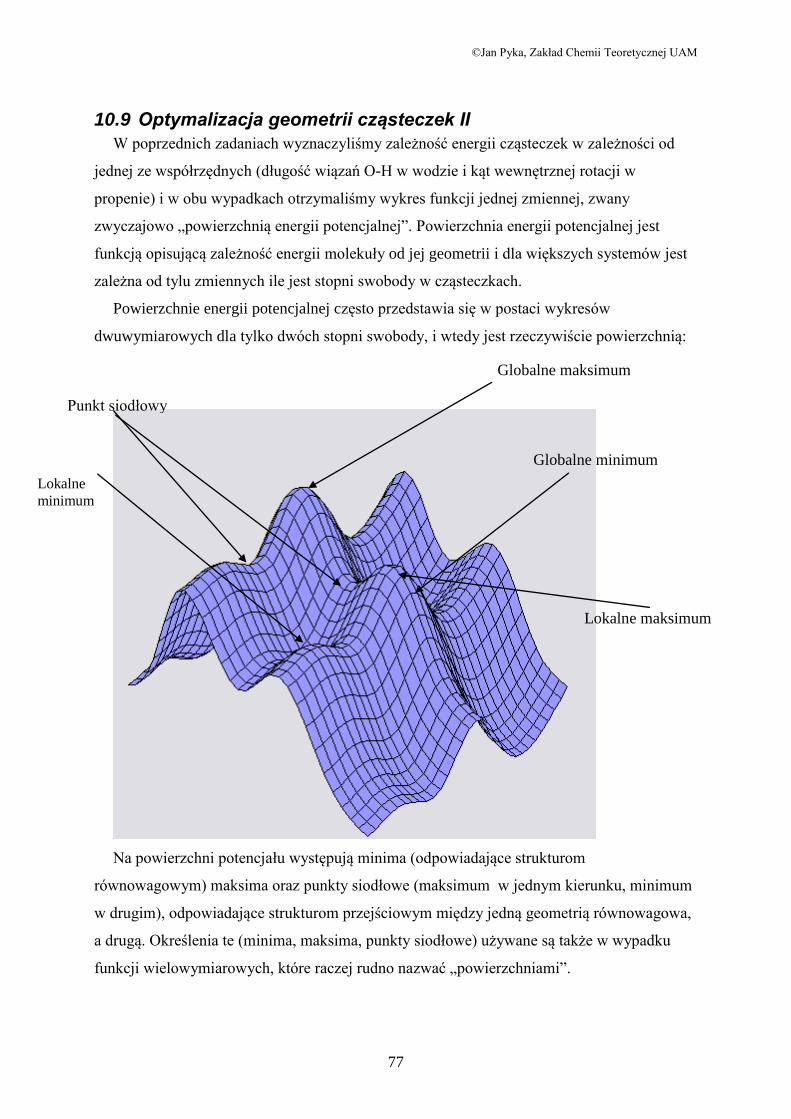
Powierzchnie energii potencjalnej często przedstawia się w postaci wykresów
dwuwymiarowych dla tylko dwóch stopni swobody, i wtedy jest rzeczywiście powierzchnią:
Na powierzchni potencjału występują minima (odpowiadające strukturom
równowagowym) maksima oraz punkty siodłowe (maksimum w jednym kierunku, minimum
w drugim), odpowiadające strukturom przejściowym między jedną geometrią równowagowa,
a drugą. Określenia te (minima, maksima, punkty siodłowe) używane są także w wypadku
funkcji wielowymiarowych, które raczej rudno nazwać „powierzchniami”.
Lokalne
minimum
Punkt siodłowy
Globalne maksimum
Globalne minimum
Lokalne maksimum

©Jan Pyka, Zakład Chemii Teoretycznej UAM
78
Optymalizując geometrię, szukamy w istocie minimów funkcji potencjału. Problem
oczywiście polega na tym, żeby właściwie określić minimum globalne, czyli geometrię
odpowiadająca najniższej energii, gdyż równie możemy trafić do któregoś z minimów
lokalnych. Wszystko zależy do tego, z jakiej struktury startujemy. Dobrze jest mniej więcej
wiedzieć, jaka jest równowagowa geometria cząsteczki i rozpoczynając obliczenia, założyć ją
w danych wejściowych.
Zadanie.1
Przeprowadzić optymalizację geometrii dla dwóch konformerów propenu, które różnią się
wartością kąta dwuściennego.
1800 0
0
Czym różnią się obie zoptymalizowane geometrie? Obliczenia należy przeprowadzić w
bazie 6-31G(d).
Uwaga: Przed ustaleniem kąta dwuściennego należy „oczyścić” strukturę przyciskiem
Clean.
Rozwiązanie
Obie optymalizacje kończą się dość szybko i każda prowadzi do innej struktury( aby się o
tym przekonać, należy wczytać oba pliki wynikowe jednocześnie i wykorzystując przycisk
Inquire porównać długości wiązań i kąty). Oznacza to, że osiągnęliśmy dwa różne punkty
stacjonarne na powierzchni energii potencjalnej (dwa minima). Gdyby istniało tylko jedno, w
obu wypadkach otrzymalibyśmy tę samą zoptymalizowana geometrię, choć w jednym
wypadku (dla geometrii wyjściowej, która znacznie by odbiegała od końcowej) oblicznei a
zapewne trwałyby znacznie dłużej.
Kąt dwuścienny Energia
00 -117,07147
1800 -117,06818

©Jan Pyka, Zakład Chemii Teoretycznej UAM
79
Widać, że między obu formami jest różnica energii około 3 milihartree. W rzeczywistości
geometria z kątem 00 reprezentuje globalne minimum, podczas gdy ta druga to minimum
lokalne. Wynika z tego, że częściowy pierścień z trzech węgli i leżącego w płaszczyźnie
jednego wodoru z grupy metylowej jest układem energetycznie nieco korzystniejszym od
drugiej formy.
Zadanie 2.
Przeprowadzić optymalizację geometrii dla dwóch konformerów alkoholu winylowego
pokazanych poniżej.
1800 0
0
Rozwiązanie.
Po optymalizacjach znowu otrzymujemy dwie różne geometrie, co łatwo sprawdzić za
pomocą przycisku Inquire. Energie zamieszczam w tabeli poniżej
Kąt dwuścienny Energia
00 -152,88889
1800 -152,88539
Widać, że podobnie jak w wypadku propenu konformer z kątem dwuściennym równym 00
ma niższą energię.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
80
10.10 Obliczenia częstości drgań
W trakcie obliczeń energii oraz optymalizacji geometrii ignoruje się wibracje systemów
molekularnych. W rzeczywistości jądra w cząsteczkach są w stałym ruchu. W stanach
równowagowych te drgania są regularne i przewidywalne, a molekuły można identyfikować
na podstawie ich charakterystycznych widm.
Gaussian może obliczyć częstości drgań i przewidzieć postać widm wibracyjnych cząstek
w ich stanach podstawowych oraz wzbudzonych. Dodatkowo GaussView został wyposażony
procedurę animacyjną, dzięki której można dosłownie zobaczyć rodzaje drgań odpowiadające
poszczególnym częstotliwościom.
Zadanie 1.
Obliczyć częstości drgań molekuły formaldehydu.
Rozwiązanie.
Obliczenia częstości drgań należy zawsze prowadzić dla punktów stacjonarnych
powierzchni energii potencjalnej. Można zrobić to w dwóch krokach:
1. Najpierw optymalizujemy geometrię.
2. Plik wynikowy z poprzedniego kroku traktujemy jako wejściowy (zmieniamy
.LOG na .gjf w nazwie) do obliczeń częstotliwości (Job Type – Frequency)
Można też połączyć oba zadania wybierając jako Job Type – Opt + Freq
Jak zwykle obliczenia przeprowadzimy bazie 6-31G(d).
Wyniki:
Po wczytaniu pliku z wynikami (.LOG), wybieramy z Menu Results pozycję Vibrations.
Ukaże się okno:

©Jan Pyka, Zakład Chemii Teoretycznej UAM
81
W pierwszych dwóch kolumnach mamy częstości drgań oraz intensywności linii. Po
naciśnięciu przycisku Spectrum program pokazuje, jak powinno wyglądać widmo w
podczerwieni. Natomiast naciśnięcie przycisku Start uruchamia wspomniana już procedurę
animacyjną, dzięki czemu możemy obejrzeć, jak wyglądają poszczególne drgania.
Uwaga: Częstości drgań obliczone na poziomie Hartree-Focka obarczone są
systematycznym błędem, który jest konsekwencją zastosowania przybliżenia
jednoelektronowego, czyli zaniedbania korelacji elektronów, przez co wyniki są zbyt duże.
Okazuje się, że można ten problem nieco zmniejszyć, jeśli wszystkie wyniku pomnożyć razy
współczynnik skalujący o wartości 0,8929 (dla bazy 6-31G(d)).
Drugą przyczyną „przestrzelenia” wyników jest to, że dane eksperymentalne reprezentują
drgania anharmoniczne, podczas gdy w obliczeniach wykorzystywany jest model drgań
harmonicznych, których częstość jest wyższa od częstości oscylacji anharmonicznych.
Zagadnieniu dokładności otrzymywanych wyników poświęcimy trochę czasu później.
Warto porównać powyższe wyniki z danymi eksperymentalnymi dla formaldehydu.
Opis drgania normalnego Eksperymentalne częstości drgań i
intensywności
Symetryczne rozciągające CH2 2783 silne
Rozciągające CO 1746 bardzo silne
Nożycowe CH2 1500 silne
Rozciągające antysymetryczne CH2 2843 bardzo silne
Wahadłowe CH2 1249 silne
Wachlarzowe CH2 1167 silne
W ramach ćwiczeń zidentyfikujcie teraz drgania.
Oczywiście zastosowanie metody bardziej zaawansowanej od HF, na przykład MP2
prowadzi do poprawy wyników, co warto sprawdzić.
Zadanie 2.
Przeprowadź obliczenia częstotliwości drgań dla molekuł z przedstawionych poniżej.
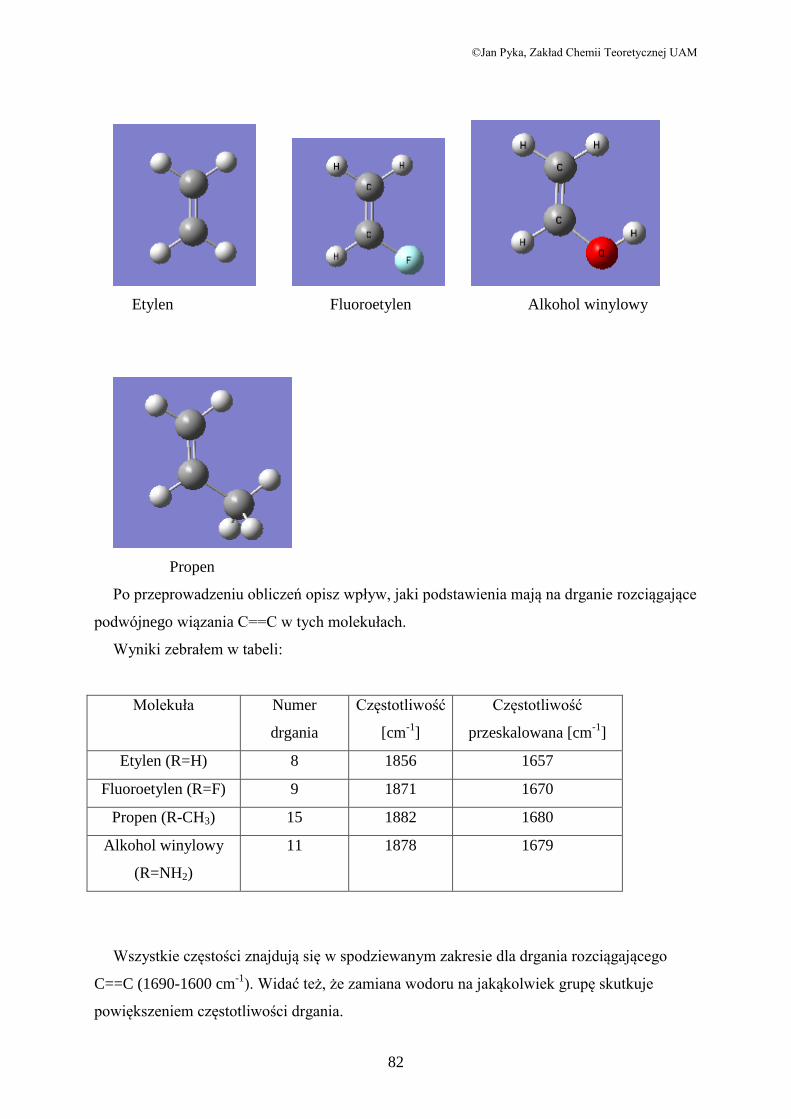
©Jan Pyka, Zakład Chemii Teoretycznej UAM
82
Etylen Fluoroetylen Alkohol winylowy
Propen
Po przeprowadzeniu obliczeń opisz wpływ, jaki podstawienia mają na drganie rozciągające
podwójnego wiązania C==C w tych molekułach.
Wyniki zebrałem w tabeli:
Molekuła Numer
drgania
Częstotliwość
[cm-1
]
Częstotliwość
przeskalowana [cm-1
]
Etylen (R=H) 8 1856 1657
Fluoroetylen (R=F) 9 1871 1670
Propen (R-CH3) 15 1882 1680
Alkohol winylowy
(R=NH2)
11 1878 1679
Wszystkie częstości znajdują się w spodziewanym zakresie dla drgania rozciągającego
C==C (1690-1600 cm-1
). Widać też, że zamiana wodoru na jakąkolwiek grupę skutkuje
powiększeniem częstotliwości drgania.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
83
10.11 Wyznaczanie geometrii kompleksów przejściowych i przewidywanie ścieżek reakcji chemicznych.
Gaussian pozwala wyznaczyć geometrię kompleksów przejściowych i przewidzieć
ścieżkę reakcji. Zwykle takie obliczenia wymagają raczej subtelnych zabiegów, my jednak
zilustrujemy je na przykładzie dosyć prostym, a mianowicie reakcji izomeryzacji cząsteczki
HCN przebiegającej zgodnie z równaniem:
Aby móc wymodelować taką reakcję należy znaleźć odpowiedni kompleks przejściowy.
Zadanie tego typu nie jest jednak problemem trywialnym. W poszukiwaniu odpowiedniego
kompleksu przejściowego możemy próbować wykorzystać wbudowana w program Gaussian
procedurę QST2. Po zdefiniowaniu w pliku wsadowym struktury początkowej oraz końcowej
program będzie próbował samodzielnie znaleźć taki kompleks przejściowy. Dowodem na
poprawność otrzymanego kompleksu będzie urojona wartość jednej i tylko jednej
częstotliwości drgań harmonicznych otrzymanego kompleksu (w GaussView urojona wartość
częstotliwości będzie przedstawiona jako ujemna).
UWAGA! Algorytmy obliczeniowe są na tyle mało skuteczne, że definiując stan
początkowy i końcowy maksymalnie zbliżony pod kątem geometrii do kompleksu
przejściowego znacznie zwiększamy swoje szanse na uzyskanie właściwego kompleksu/
W podobny sposób postąpimy i w tym ćwiczeniu. Optymalizacja geometrii cząsteczki
HCN prowadzi zawsze do liniowej formy typu H-C≡N. Aby uzyskać właściwy wynik, w
obliczeniach przyjmiemy dla początkowej struktury wartość kąta HCN równą 150º. Podobnie
definiując geometrię cząsteczki CNH, ustawimy początkową wartość kąta CNH na 150º.
W tym celu
1. Uruchomiamy program GaussView
2. Po dwukrotnym kliknięciu ikony R-Group Fragment z okna Select R-Group
Fragment wybieramy grupę cyjanową (cyano).
3. Z okna Current Fragment wklejamy cząsteczkę HCN do okna G1:M1:V1-New
4. Klikamy na ikonę Modify Angle, a potem kolejno na atomy wodoru, węgla i
azotu w oknie G1:M1:V1-New, i zmieniamy wartość kąta płaskiego HCN ze 180º
na 150º

©Jan Pyka, Zakład Chemii Teoretycznej UAM
84
5. W menu głównym wybieramy File/New/Add to MolGroup
6. W nowym oknie G1:M2:V1-New konstruujemy cząsteczkę C≡NH. W tym celu
możemy ponownie skorzystać z gotowej cząsteczki HCN. Po usunięciu atomu
wodoru wklejamy go po stronie atomu azotu. Na pasku narzędzi klikamy na ikonę
Modify Bond, a następnie na atom azotu oraz atom węgla. Ustawiamy rodzaj
wiązania na pojedyncze oraz jego długość na 1,2 Å. Zmieniamy także (funkcja
Modify Angle) wartość kąta płaskiego na 150º.
7. Zapisujemy plik we właściwym folderze pod nazwą HCN1.GJF
8. W menu głównym wybieramy Calculate/Gaussian
9. W zakładce Job Type wybieramy Optimization, w polu Optimize to a parameter
TS(QST2), natomiast poniżej w polu Calculate Force Constants opcję Always
10. W zakładce Link0 klikamy na przycisk Checkpoint File i zapisujemy plik typu
checkpoint pod nazwą HCN1.CHK
11. Klikamy na przycisk Submit
12. Po ukończeniu obliczeń otwieramy plik wynikowy HCN1.LOG
Sprawdzamy poprawność otrzymanego kompleksu przejściowego optymalizując jego
geometrię oraz badając częstotliwość jego drgań normalnych. W tym celu po otwarciu pliku
HCN1.LOG
1. Klikamy na zakładkę Calculate/Gaussian
2. W zakładce Job Type wybieramy Opt+Freq, w polu Optimize to a parametr
TS(Berry), natomiast poniżej, w polu Calculate Force Constants opcję Always
3. W zakładce Link0 klikamy przycisk Checkpoint File i zapisujemy plik typu
checkpoint pod nazwą HCN2.CHK
4. Klikamy na przycisk Submit
5. Zapisujemy plik pod nazwą HCN2.GJF
6. Po ukończeniu obliczeń otwieramy plik wynikowy HCN2.LOG

©Jan Pyka, Zakład Chemii Teoretycznej UAM
85
7. Sprawdzamy częstotliwość drgań normalnych. Jeśli jedna i tylko jedna
częstotliwość ma wartość urojoną (program przedstawia urojoną częstotliwość
jako ujemną), możemy oczekiwać, że otrzymaliśmy poprawny kompleks
przejściowy.
8. Zamykamy plik HCN2.LOG, a następnie otwieramy plik HCN2.CHK
9. Klikamy na zakładkę Calculate/Gaussian
10. W zakładce Job Type wybieramy IRC (intrinsic reaction coordinate –
współrzędna reakcji), w polu Follow IRC wybieramy Both directions, a w polu
Compute more points, N= wpisujemy wartość 30

©Jan Pyka, Zakład Chemii Teoretycznej UAM
86
11. W zakładce Link0 klikamy przycisk Checkpoint File i zapisujemy plik typu
checkpoint pod nazwą HCN3.CHK
12. Klikamy na przycisk Submit
13. Zapisujemy plik wejściowy pod nazwą HCN3.GJF
14. Po ukończeniu obliczeń otwieramy plik wynikowy HCN3.CHK
15. Z menu głównego wybieramy Results/IRC
Po naciśnięciu na zielone kółeczko zobaczymy animację przebiegu reakcji.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
87
11 Ograniczenia metody Hartree-Focka Metoda Hartree-Focka pozwala uzyskać przybliżone wyniki dla wielu parametrów
molekularnych i jest przez to bardzo użyteczna. Jednak czasem te przybliżone wyniki dość
znacznie odbiegają od danych eksperymentalnych i dlatego w dokładniejszych obliczeniach
traktuje się je wstępne, wykorzystywane następnie w metodach bardziej zaawansowanych.
Jedną z tych bardziej zaawansowanych metod jest rachunek zaburzeń Møllera-Plesseta. W
zależności od tego, na której poprawce tego rachunku kończymy obliczenia, mówimy o
metodzie MP2, MP3, lub MP4.
Fluorowodór
Istnieje wiele molekuł, dla których uwzględnienie korelacji elektronowej (wyjście poza
jednowyznacznikową funkcję elektronową) jest konieczne dla uzyskania w miarę dokładnych
wyników. Jednym z najprostszych jest fluorowodór. Policzymy teraz energię wiązania
fluorowodoru, posługując się różnymi metodami. Chcąc znaleźć energię wiązania
fluorowodoru, obliczymy najpierw energie atomu fluoru i atomu wodoru, a następnie od
sumy tych energii odejmiemy energię fluorowodoru.
W poniższej tabeli znajdziecie przewidywane energie wiązania cząsteczki fluorowodoru
obliczone różnymi metodami w bazie 6-311++G(3df,3pd). Wymieramy tę bazę, ponieważ jest
ona największa i powinna dać najlepsze wyniki. Warto także najpierw przeprowadzić
optymalizację cząsteczki (np. metodzie HF) i zoptymalizowaną geometrię wykorzystywać w
obliczeniach bardziej dokładnymi metodami – robi się bardzo prosto: wczytujemy plik
wynikowy z optymalizacji i zmieniwszy tylko metodę (np. z HF na MP2) obliczamy energię.
Uwaga: GaussView umożliwia nam odczytanie energii dla HF oraz MP2. jeśli chcemy
sprawdzić wynik, który otrzymaliśmy metodą MP4 lub QCISD musimy otworzyć plik
Gaussiana (Na dole Menu Results jest opcja View File) Jest on trochę deprymujący,
(przynajmniej na początku), ale na szczęście to, co nas interesuje, jest na samym końcu. Dla
metody MP4 wygląda to na przykład tak:
Symmetry A1 KE= 8.744159201066D+01 Symmetry A2 KE=-1.935053868449D-51 Symmetry B1 KE= 6.269162722288D+00 Symmetry B2 KE= 6.269162722288D+00 1|1|UNPC-UNK|SP|RMP4SDTQ-FC|6-311++G(3df,3pd)|F1H1|PCUSER|10-Jan-2010| 0||# MP4(SDTQ)/6-311++G(3DF,3PD) GEOM=CONNECTIVITY||Title Card Require d||0,1|F|H,1,0.916659||Version=x86-Win32-G03RevB.03|State=1-SG|HF=-100 .0576469|MP2=-100.33282|MP3=-100.3318454|MP4D=-100.3357761|MP4DQ=-100. 3328491|MP4SDQ=-100.3349599|MP4SDTQ=-100.3430608|RMSD=4.859e-009|PG=C* V [C*(H1F1)]||@
E(MP2) E(MP3)
E(MP4)
©Jan Pyka, Zakład Chemii Teoretycznej UAM
88
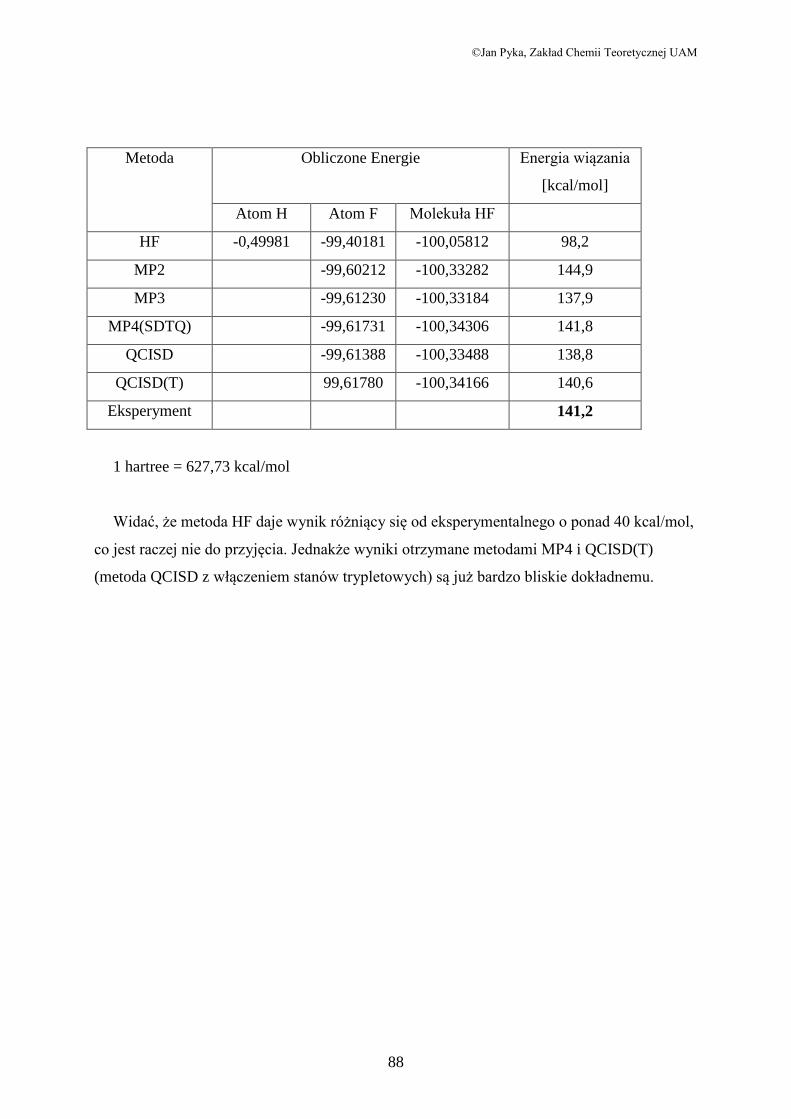
Metoda Obliczone Energie Energia wiązania
[kcal/mol]
Atom H Atom F Molekuła HF
HF -0,49981 -99,40181 -100,05812 98,2
MP2 -99,60212 -100,33282 144,9
MP3 -99,61230 -100,33184 137,9
MP4(SDTQ) -99,61731 -100,34306 141,8
QCISD -99,61388 -100,33488 138,8
QCISD(T) 99,61780 -100,34166 140,6
Eksperyment 141,2
1 hartree = 627,73 kcal/mol
Widać, że metoda HF daje wynik różniący się od eksperymentalnego o ponad 40 kcal/mol,
co jest raczej nie do przyjęcia. Jednakże wyniki otrzymane metodami MP4 i QCISD(T)
(metoda QCISD z włączeniem stanów trypletowych) są już bardzo bliskie dokładnemu.

©Jan Pyka, Zakład Chemii Teoretycznej UAM
89
Spis Treści
1 Podstawowe funkcje programu Gaussview/Gaussian ........................................................ 1 2 GaussView ......................................................................................................................... 3
2.1 Ustawienia opcji wyświetlania i obracanie cząsteczki ................................................ 3 2.2 Wybór fragmentu ......................................................................................................... 6
2.3 Wybór grupy. ............................................................................................................... 7 2.4 Dodawanie pojedynczych aminokwasów lub nukleotydów ........................................ 7 2.5 Dodawanie oraz edycja własnych fragmentów cząsteczek do biblioteki .................... 7 2.6 Opcja „HOT” Atom. .................................................................................................... 8
2.7 Wizualizacja cząsteczek ............................................................................................ 10 2.8 Zmiana odległości między atomami .......................................................................... 11 2.9 Zmiana kąta między atomami .................................................................................... 13
2.10 Zmiana kąta dwuściennego .................................................................................... 13 2.11 Odczytywanie (a nie modyfikowanie) wartości parametrów geometrycznych ..... 15 2.12 Usuwanie atomów oraz dodawanie atomów wodoru ............................................ 15 2.13 Reszta przycisków z Konstruktora ......................................................................... 15
3 Używanie różnych menu programu GaussView .............................................................. 17 3.1 Opcje menu File ......................................................................................................... 17
3.1.1 New: Tworzenie nowego modelu ...................................................................... 17 3.1.2 Open: Otwieranie plików. .................................................................................. 17 3.1.3 Save: zapisywanie plików .................................................................................. 17
4 Budowanie molekuł: przykłady ....................................................................................... 18
4.1 Pirydyna ..................................................................................................................... 18 4.2 Fenylopirydyna .......................................................................................................... 18 4.3 Purpura tyryjska ......................................................................................................... 19
4.4 Łączenie dwóch struktur: budowa dwuchlorodifenylu ............................................. 22 5 Ustawianie ładunku cząsteczki kationy, aniony ............................................................... 25
6 Przygotowanie zadania obliczeniowego .......................................................................... 26 6.1 Ustawienia parametrów obliczeń ............................................................................... 26 6.2 Wybór rodzaju zadania i metody obliczeń ................................................................ 27
6.3 Plik typu checkpoint .................................................................................................. 28 6.4 Zapisywanie pliku do obliczeń .................................................................................. 29
7 Przykład obliczeń ............................................................................................................. 30
8 Obrazowanie orbitali molekularnych, gęstości elektronowych i potencjałów
elektrostatycznych molekuł. ..................................................................................................... 36
8.1 Orbitale molekularne ................................................................................................. 36 8.2 Gęstość elektronowa .................................................................................................. 40
8.3 Potencjał elektrostatyczny molekuły ......................................................................... 43 9 Odrobina Teorii ................................................................................................................ 47
9.1 Metoda Hartree-Focka ............................................................................................... 47
9.2 Bazy Orbitali Atomowych ......................................................................................... 48 9.2.1 Orbitale slaterowskie (STO –Slater Type Orbitals) ........................................... 48
9.2.2 Orbitale gaussowskie – GTO (Gaussian Type Orbitals) .................................... 49 9.3 Orbitale obsadzone i wirtualne .................................................................................. 50 9.4 Ograniczona i nieograniczona metoda Hartree-Focka .............................................. 51
10 Ćwiczenia ......................................................................................................................... 53 10.1 Energia stanu podstawowego atomu wodoru......................................................... 53

©Jan Pyka, Zakład Chemii Teoretycznej UAM
90
10.2 Energia stanu podstawowego i orbitale atomowe atomu węgla. ........................... 54 10.3 Konfiguracje elektronowe potasu, wapnia, skandu i tytanu. ................................. 57 10.4 Energie stanu podstawowego atomów i jonów pierwszego szeregu ..................... 58 10.5 Badania molekuł .................................................................................................... 59
10.5.1 Obliczanie energii dla określonej geometrii cząsteczki (single point energy) ... 59 10.6 „Ręczna” Optymalizacja długości wiązań dla cząsteczki wody ............................ 65 10.7 Wykres (scan) energii potencjalnej ........................................................................ 67 10.8 Wyznaczenie optymalnej (równowagowej) geometrii molekuły wody pomocą
Gaussiana ............................................................................................................................. 72
10.9 Optymalizacja geometrii cząsteczek II .................................................................. 77 10.10 Obliczenia częstości drgań ..................................................................................... 80 10.11 Wyznaczanie geometrii kompleksów przejściowych i przewidywanie ścieżek
reakcji chemicznych. ............................................................................................................ 83 11 Ograniczenia metody Hartree-Focka ............................................................................... 87 Spis Treści ................................................................................................................................ 89


















