
1 Estimating chromosomal copy number InCoB2007, Hong Kong, 30 August, 2007.
50
1 Estimating chromosomal copy number InCoB2007, Hong Kong, 30 August, 2007
-
Upload
morgan-farmer -
Category
Documents
-
view
223 -
download
0
Transcript of 1 Estimating chromosomal copy number InCoB2007, Hong Kong, 30 August, 2007.

1
Estimating chromosomal copy number
InCoB2007, Hong Kong, 30 August, 2007

2
Copy number variation (CNV)What is it?
• A form of human genetic variation: instead of 2 copies of each region of each chromosome (diploid), some people have amplifications or losses (> 1kb) in different regions– this doesn’t include translocations or inversions
• We all have such regions – the publicly available genome NA15510 has between 5 & 240 by various estimates
– they are only rarely harmful (but rare things do happen)
3
Copy number variationPopulation genomics
The genomes of two humans differ more in a structural sense than at the nucleotide level; a recent paper estimates that on average two of us differ by
~ 4 - 24 Mb of genetic due to Copy Number Variation
~ 2.5 Mb due to Single Nucleotide Polymorphisms

4
Copy number variationAs it relates to human disease
Is responsible for a number of rare genetic conditions. For example, Down syndrome ( trisomy 21), Cri du chat syndrome (a partial deletion of 5p).
Is implicated in complex diseases. For example: CCL3L1 CN HIV/AIDS susceptibility; also, some sporadic (non-inherited) CN variants are strongly associated with autism, while
Tumors typically have a lot of chromosomal abnormalities, including recurrent CN changes.
5
Trisomy 21
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

6
Partial deletion of chr 5p
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.

7
Large amplifications/losses can be seen by eye; smaller ones
are hard to see

8
A cytogeneticist’s story
“The story is about diagnosis of a 3 month old baby with macrocephaly and some heart problems. The doctors questioned a couple of syndromes which we tested for and found negative. Rather than continue this ‘shot in the dark’ approach, we put the case on an array and found a 2Mb deletion which notably deletes the gene NSD1 on chr 5, mutations in which are known to be cause Sotos syndrome. This is an overgrowth syndrome and fits with the macrocephaly.
The bottom line is that we are able to diagnose quicker by this approach and delineate exactly the underlying genetic change.”

9
2Mb deletion
Chromosome 5
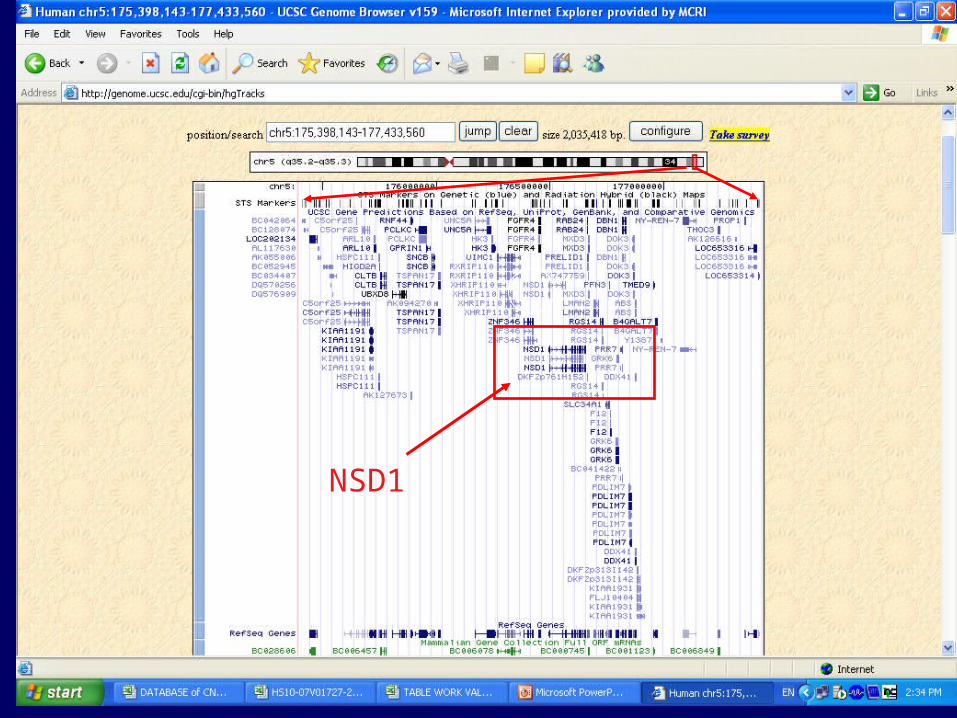
10
NSD1

11A lung cancer cell line vs matched normal lymphoblast,from Nannya et al Cancer Res 2005;65:6071-6079
Many tumors have gross CN changes

12
Research into gonad dysfunction: Human sex reversal
• 20% of 46,XY females have mutations in SRY
• 80% of 46,XY females unexplained!
• 90% of 46,XX males due to translocation SRY
• 10% of 46,XX males unexplained!
Suggests loss of function and gain of function mutations in other genes may cause sex reversal. We’re looking at shared deletions.

13
Plan
To introduce the Single Nucleotide Polymorphism (SNP) arrays, the probes, and the associated assays. Then I’ll discuss the first bioinformatic aspect of Copy Number (CN) analysis, what I call low-level analyses, then show one way of assessing the outcome.
For simplicity I concentrate on Affymetrix arrays, called GeneChips, though similar considerations apply in whole or in part to some other array technologies, including Illumina.

14
Genomic DNA
ATCGGTAGCCATTCATGAGTTACTAPerfect Match probe for Allele A
ATCGGTAGCCATCCATGAGTTACTAPerfect Match probe for Allele B
A SNP
GTAGCCATCGGTA GTACTCAATGAT
Affymetrix SNP chip terminology
Genotyping: answering the question about the two copies of the chromosome on which the SNP is located:
Is a sample AA (AA) , AB (AG) or BB (GG) at this SNP?
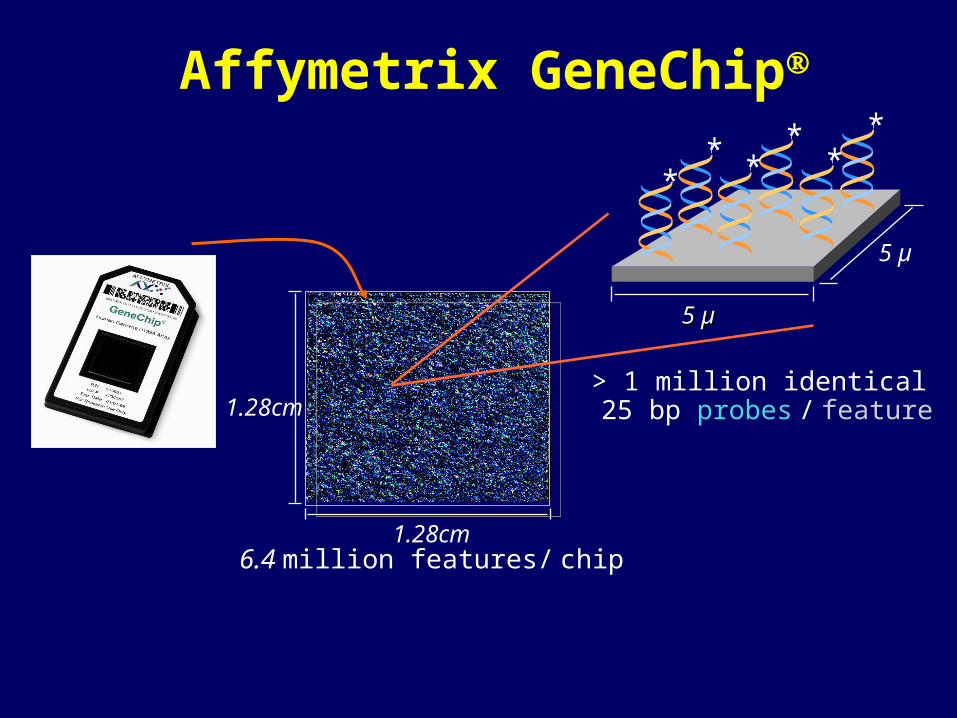
Affymetrix GeneChip
1.28cm 6.4 million features/ chip
1.28cm
5 µ5 µ
5 µ
> 1 million identical 25 bp probes / feature
* **
***
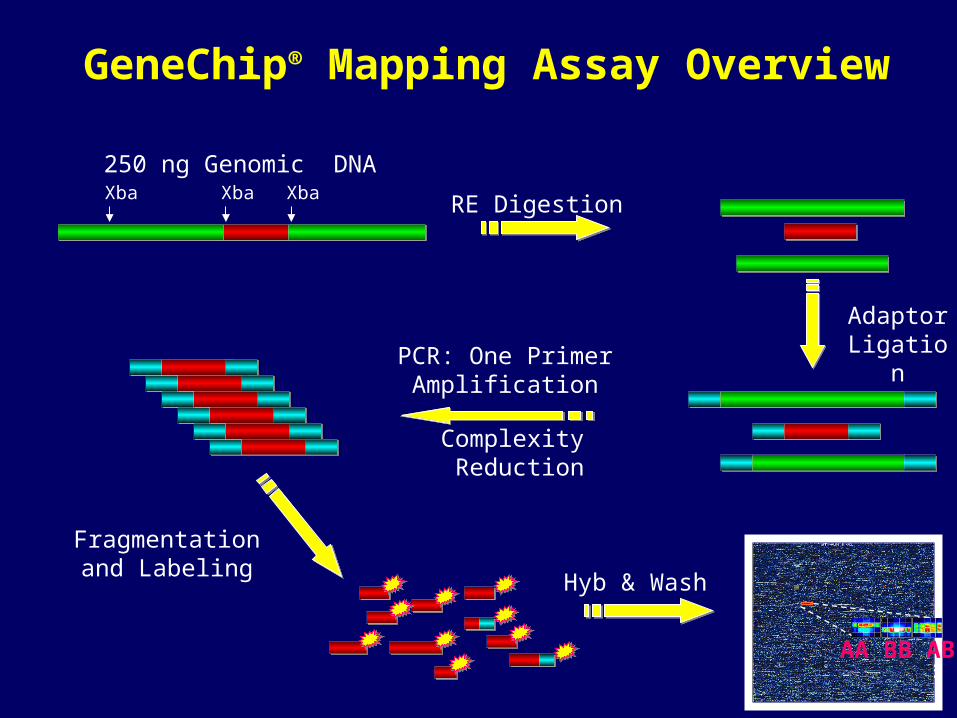
16
250 ng Genomic DNA
RE Digestion
Adaptor Ligation
GeneChip® Mapping Assay Overview
Xba XbaXba
Fragmentationand Labeling
PCR: One Primer Amplification
Complexity Reduction
AA BB AB
Hyb & Wash

17
Principal low-level analysis steps
• Background adjustment and normalization at probe level These steps are to remove lab/operator/reagent effects
• Combining probe level summaries to probe set level summary: best done robustly, on many chips at once
This is to remove probe affinity effects and discordant observations (gross errors/non-responding probes, etc)
• Possibly further rounds of normalization (probe set level)
as lab/cohort/batch/other effects are frequently still visible
• Derive the relevant copy-number quantities Finally, quality assessment is an important low-level task.
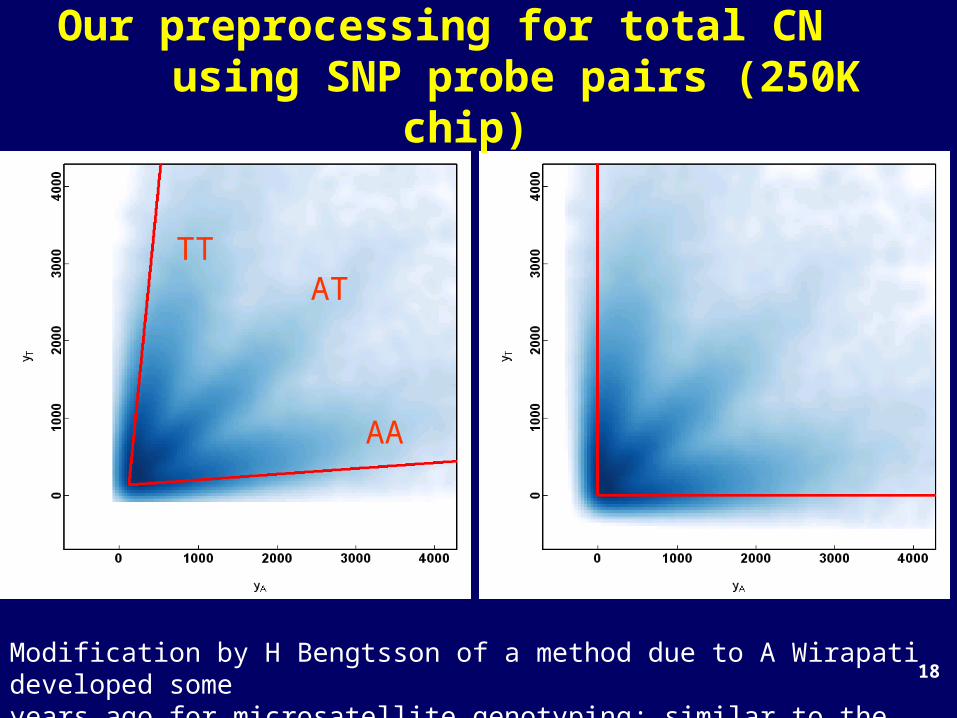
18
AA
TTAT
Our preprocessing for total CN using SNP probe pairs (250K
chip)
Modification by H Bengtsson of a method due to A Wirapati developed some years ago for microsatellite genotyping; similar to the approach used by Illumina.
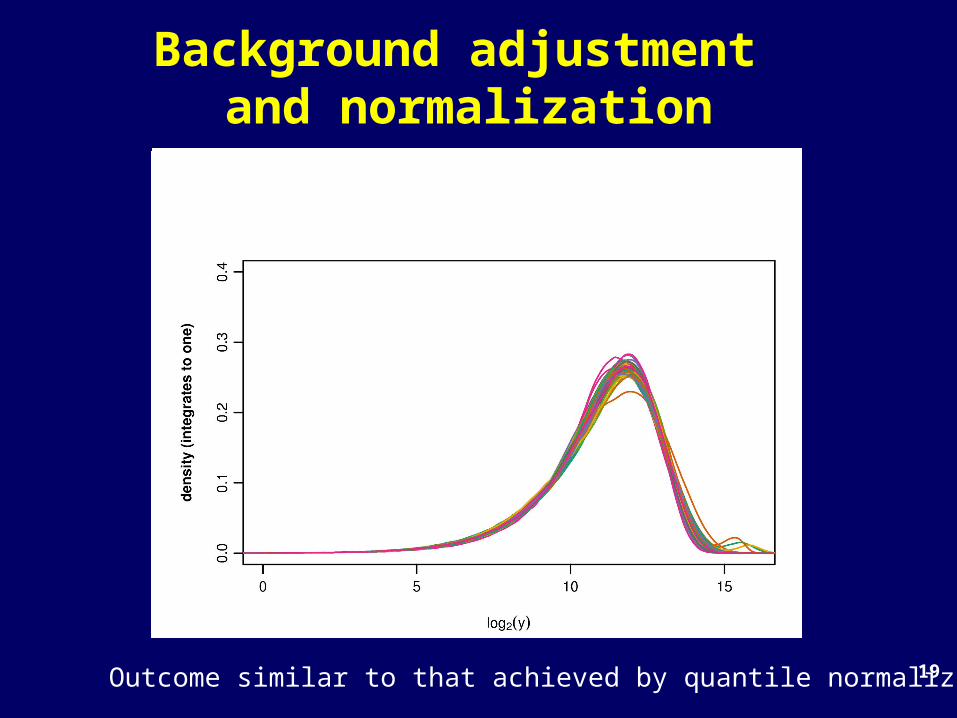
19
Background adjustment and normalization
Outcome similar to that achieved by quantile normalization

20
Low-level analysis problems haven’t been solved once and
for all; why?
• The feature size keeps and so the # features/chip keeps;
• Fewer and fewer features are used for a given measurement, allowing more measurements to be made using a single chip
These considerations all place more and more demands on the low-level analysis: to maintain the quality of existing measurements, and to obtain good new ones.

21
SNP probe tiling strategy
TAGCCATCGGTA N
SNP 0 position
A / G
GTACTCAATGAT*
ATCGGTAGCCAT T
ATCGGTAGCCAT CATCGGTAGCCAT G
ATCGGTAGCCAT ACATGAGTTACTACATGAGTTACTA
CATGAGTTACTA CATGAGTTACTA
PMMM
PMMM
AA
B B
0 Allele0 Allele
0 Allele0 Allele
Central probe quartet

22
SNP probe tiling strategy, 2
TAGCCATCGGTA N
SNP
+4 PositionA / G
GTA C TCAATGATCAGCT*
GTAGCCAT T
GTAGCCAT CGTAGCCAT C
GTAGCCAT TCAT G AGTTACTAGTCGCAT C AGTTACTAGTCG
CAT G AGTTACTAGTCGCAT C AGTTACTAGTCG
PMMM
PMMM
AA
B B
+4 Allele+4 Allele
+4 Allele+4 Allele
+4 offset probe quartet

SNP probe tiling strategy, 3
1 2 3 4 5 6 7
PMA PMA PMA PMA PMA PMA PMA
MMA MMA MMA MMA MMA MMA MMA
PMB PMB PMB PMB PMB PMB PMB
MMB MMB MMB MMB MMB MMB MMB
Central quartet
Offset quartets Offset quartets
This was repeated on the opposite strand giving 56 probes for the 10K chip.
The 100K chip had 40 chosen from offsets and strands by performance.
The 5.0 chip had 8 well chosen probes/SNP; no MMs.
The current 6.0 chip has just 6: 3 replicates of a PMA and 3 of a PMB. Also,
there are a large # of unreplicated non-polymorphic probes for CN inference.

24
What comes next?
• Using SNP chips to identify change in total copy number (i.e. CN ≠ 2)
• Outline a new method (CRMA)
• Evaluate and compare it with other methods
• Make some closing remarks on further issues

25
Copy-number estimation using Robust Multichip Analysis (CRMA)
CRMA
Preprocessing(probe signals)
allelic crosstalk (or
quantile)
Total CN PM=PMA+PMB
Summarization (SNP signals )
log-additivePM only
Post-processing fragment-length(GC-content)
Raw total CNs R = Reference
Mij = log2(ij/Rj) chip
i, probe j
A few details are passed over. Ask me later if you care about them.

26
CRMA, 1
CRMA
Preprocessing(probe signals)
allelic crosstalk (or
quantile)
Total CN PM=PMA+PMB
Summarization (SNP signals )
log-additivePM only
Postprocessing fragment-length(GC-content)
Raw total CNs Mij = log2(ij/Rj)
Already briefly described.

27
CRMA, 2
CRMA
Preprocessing(probe signals)
allelic crosstalk (quantile)
Total CN PM=PMA+PMB
Summarization (SNPsignals )
log-additivePM only
Postprocessing
fragment-length(GC-content)
Raw total CNs
Mij = log2(ij/Rj)
That’s it!

28
CRMA, 3
CRMA
Preprocessing(probe signals)
allelic crosstalk (quantile)
Total CNs PM=PMA+PMB
Summarization (SNP signals )
log-additivePM only
Postprocessing
fragment-length(GC-content)
Raw total CNs
Mij = log2(ij/Rj)
log2(PMijk) = log2ij + log2jk + ijk
Fit using rlm

29
CRMA, 4a
CRMA
Preprocessing(probe signals)
allelic crosstalk (quantile)
Total CN PM=PMA+PMB
Summarization (SNP signals )
log-additivePM-only
Postprocessing
fragment-length
(GC-content)
Raw total CNs
Mij = log2(ij/Rj)
100K
Longer fragments get lesswell amplified by PCR and so give weaker SNP signals
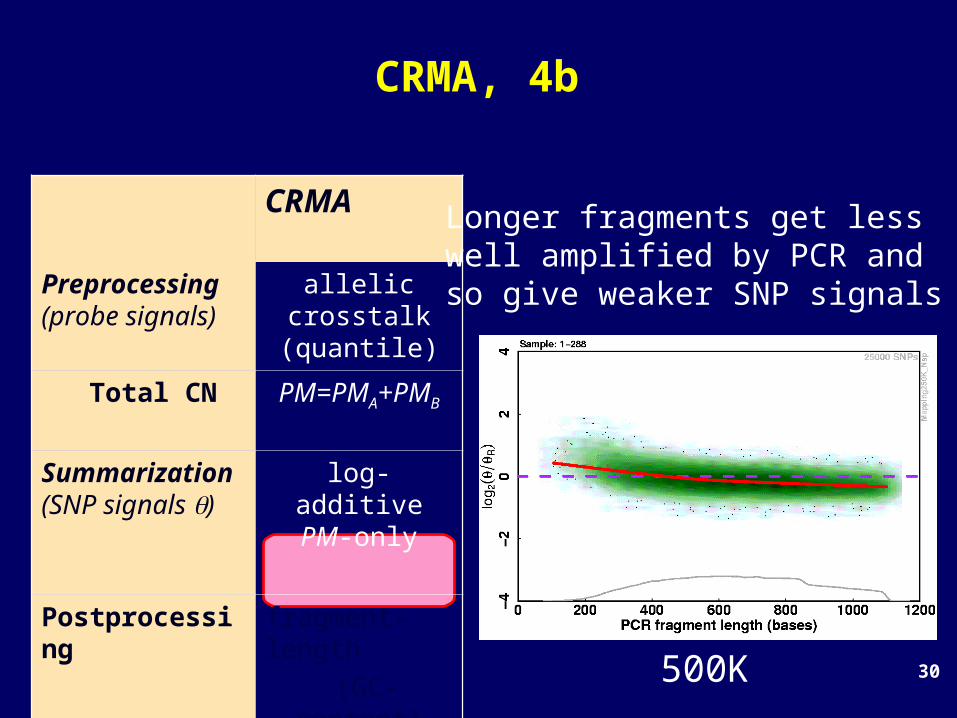
30
CRMA, 4b
CRMA
Preprocessing(probe signals)
allelic crosstalk (quantile)
Total CN PM=PMA+PMB
Summarization (SNP signals )
log-additivePM-only
Postprocessing
fragment-length
(GC-content)
Raw total CNs
Mij = log2(ij/Rj)
500K
Longer fragments get lesswell amplified by PCR and so give weaker SNP signals
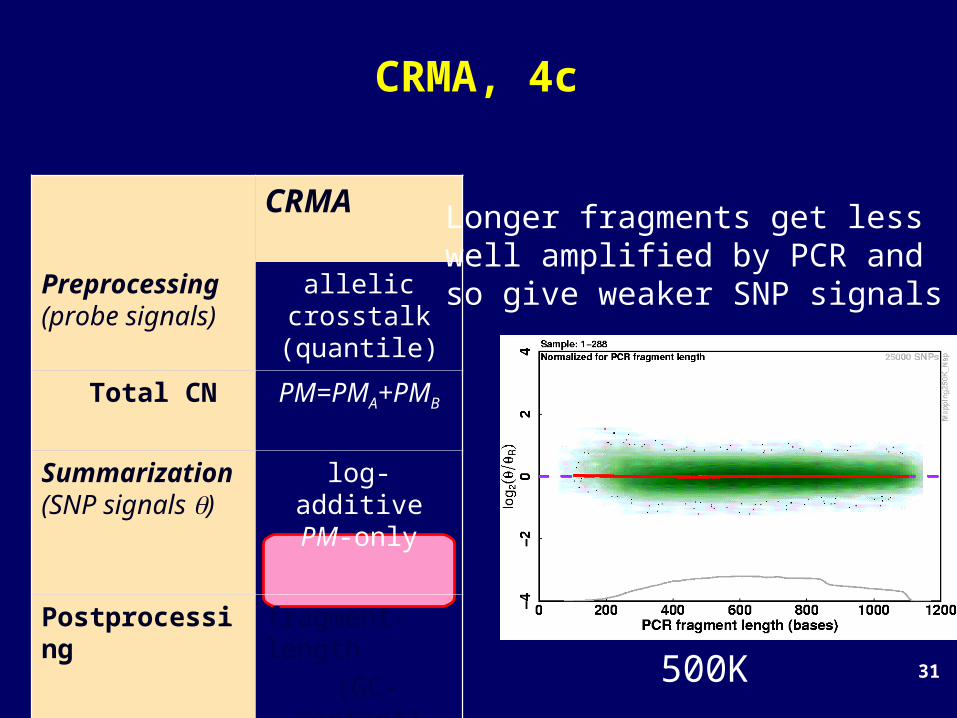
31
CRMA, 4c
CRMA
Preprocessing(probe signals)
allelic crosstalk (quantile)
Total CN PM=PMA+PMB
Summarization (SNP signals )
log-additivePM-only
Postprocessing
fragment-length
(GC-content)
Raw total CNs
Mij = log2(ij/Rj)
500K
Longer fragments get lesswell amplified by PCR and so give weaker SNP signals

32
CRMA, 5
CRMA
Preprocessing(probe signals)
allelic crosstalk (quantile)
Total CN PM=PMA+PMB
Summarization (SNP signals )
log-additivePM-only
Postprocessing
fragment-length
(GC-content)
Raw total CNs Mij = log2(ij/Rj)
Care required with the number and nature of Reference samples used
33
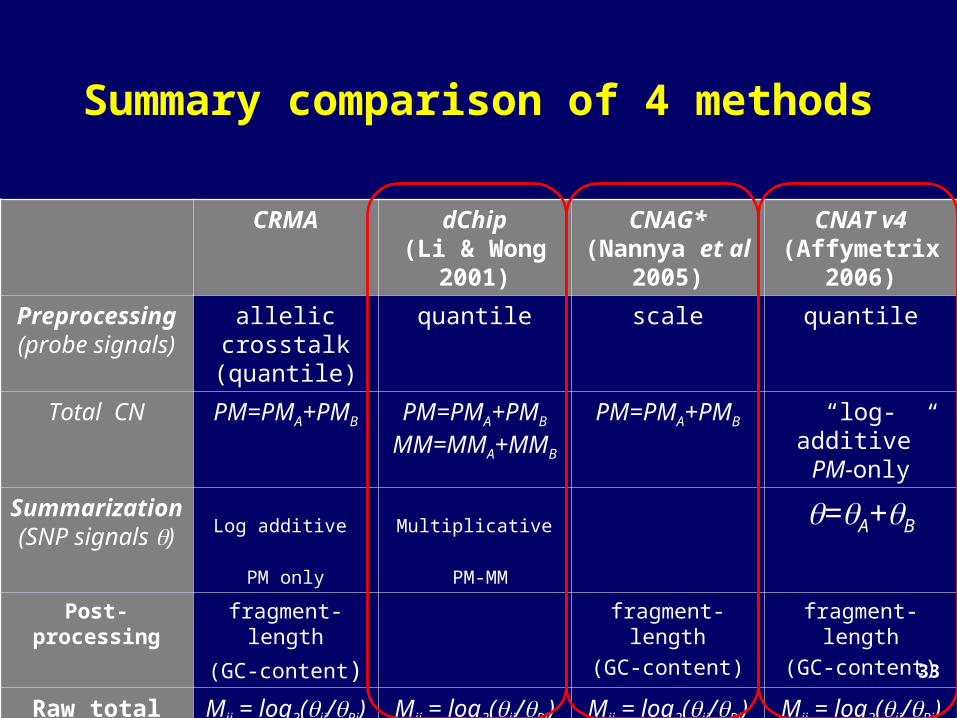
Summary comparison of 4 methods
CRMA dChip(Li & Wong
2001)
CNAG*(Nannya et al 2005)
CNAT v4(Affymetrix
2006)
Preprocessing
(probe signals)
allelic crosstalk (quantile)
quantile scale quantile
Total CN PM=PMA+PMB PM=PMA+PMB
MM=MMA+MMB
PM=PMA+PMB “log-additive”PM-only
Summarization (SNP
signals )
Log additive
PM only
Multiplicative
PM-MM
=A+B
Post-processing
fragment-length
(GC-content)
fragment-length
(GC-content)
fragment-length
(GC-content)
Raw total CNs
Mij = log2(ij/Rj)
Mij = log2(ij/Rj)
Mij = log2(ij/Rj)
Mij = log2(ij/Rj)

34
Evaluation: how well can we differentiate
between one and two copies?HapMap:Mapping 250K Nsp data30 males and 29 females (no children; one bad data set)
Chromosome X is known: Males (CN=1) & females (CN=2)5,608 SNPs
Classification rule:Mij < threshold CNij =1, otherwise CNij =2.
Number of calls: 595,608 = 330,872
35
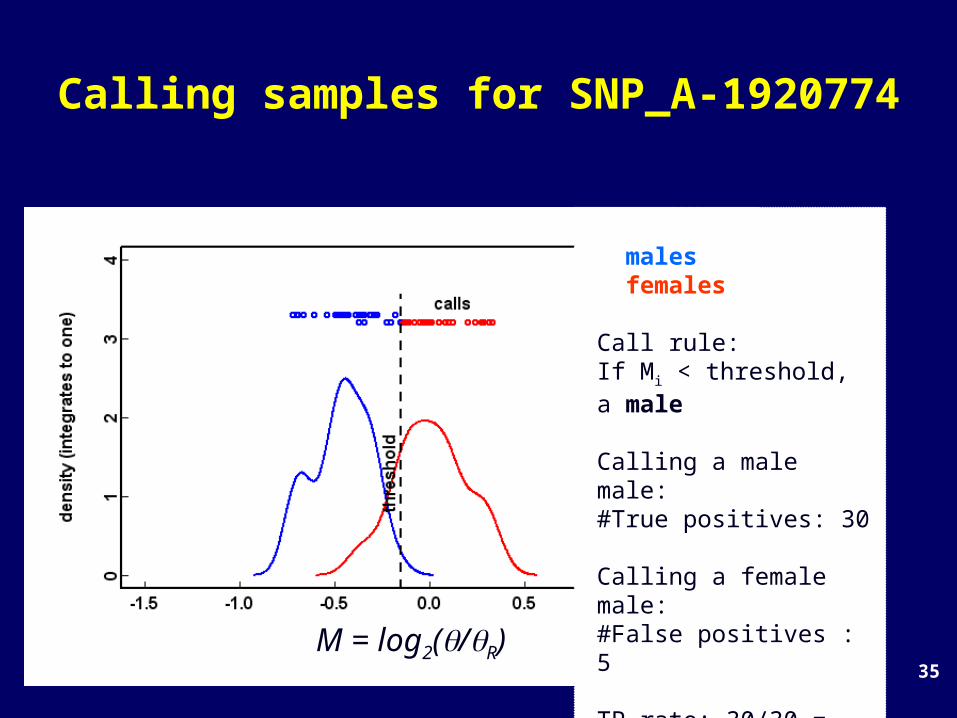
Calling samples for SNP_A-1920774
# males: 30# females: 29
Call rule:If Mi < threshold, a male
Calling a male male:#True positives: 30
Calling a female male:#False positives : 5
TP rate: 30/30 = 100%FP rate: 5/29 = 17%M = log2(/R)

36
Receiver Operator Characteristic (ROC)
increasingthreshold
FP rate
TP
rate
37
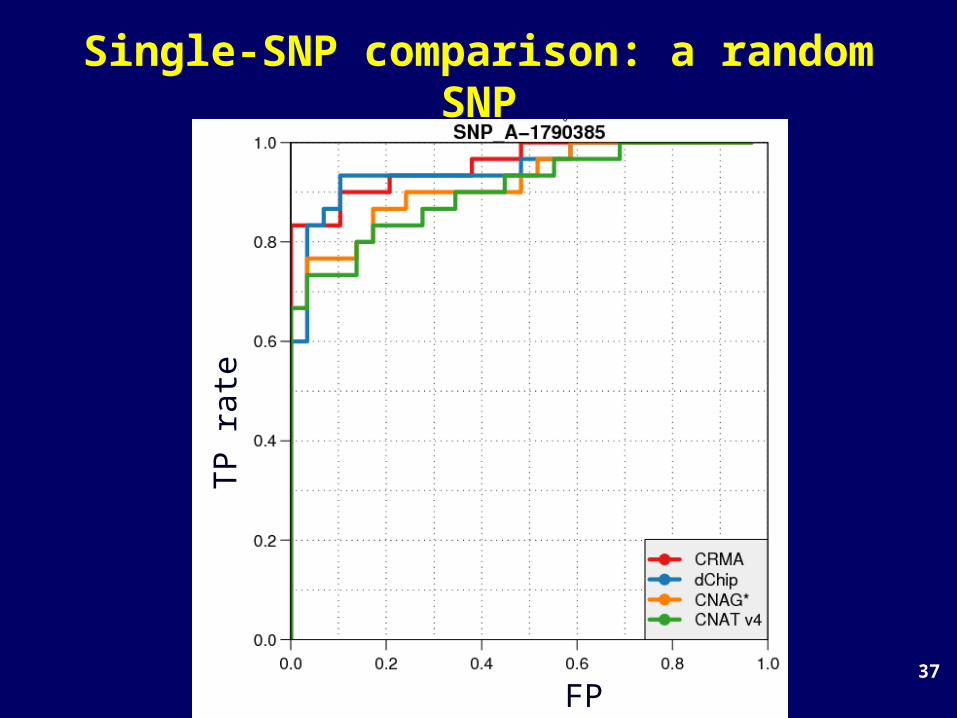
Single-SNP comparison: a random SNP
TP rate
FP rate
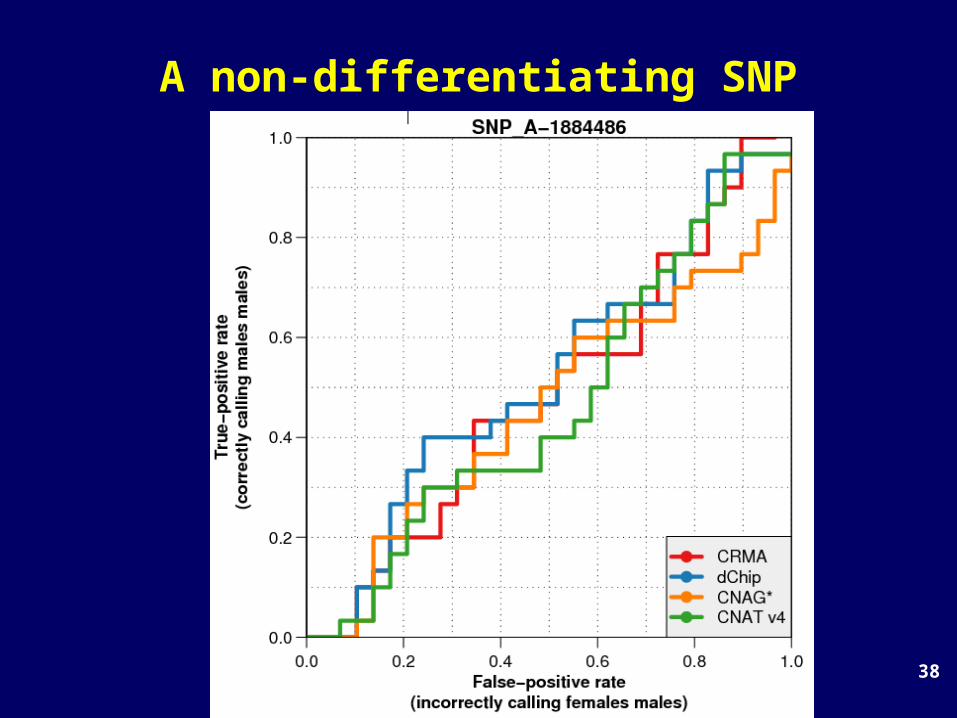
38
A non-differentiating SNP

39
Distribution (density) of TP rates when
controlling for FP rate (5,608 SNPs)
TP rate(correctly calling males male)
FP rate: 1.0% (incorrectly calling females male)
CNAT: 10% SNPs poor
density

40
CRMA & dChip perform better for an average SNP (common
threshold)Number of calls:595,608 = 330,872
zoom in
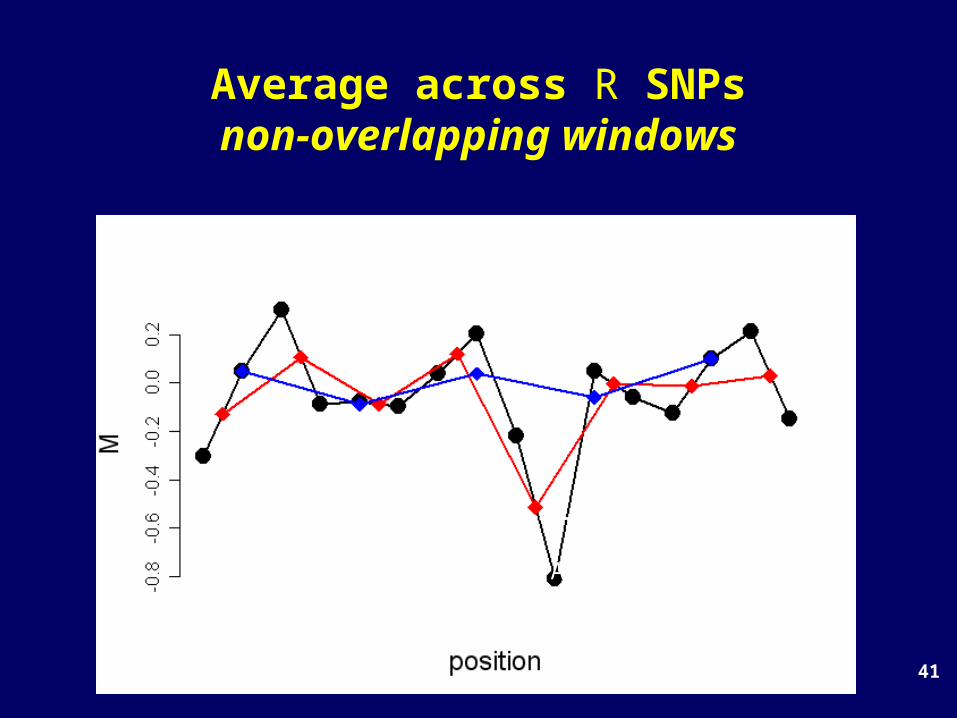
41
Average across R SNPsnon-overlapping windows
threshold
A false-positive(or real?!?)

42
Better detection rate when averaging
(with risk of missing short regions)
R=1(no av)
R=2
R=3
R=4
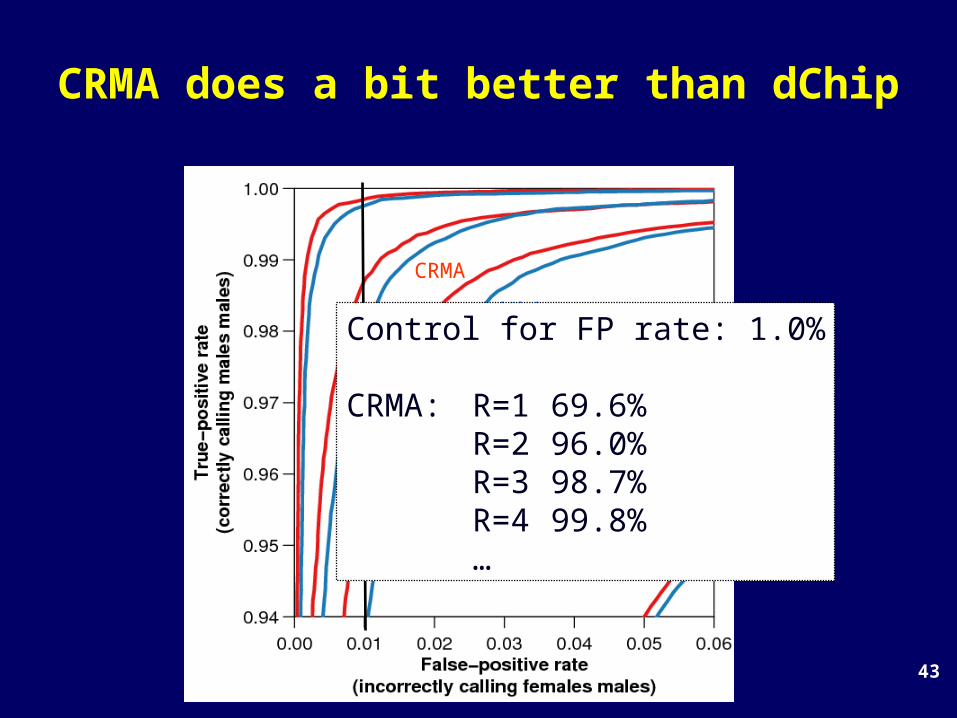
43
CRMA does a bit better than dChip
CRMA
dChipControl for FP rate: 1.0%
CRMA: R=1 69.6%R=2 96.0%R=3 98.7%R=4 99.8%…

44
Comparing methods by “resolution”
CRMAdChip
CNAG*
CNAT
All @ FP rate 1%

45
Several further bioinformatic issues
Estimating copy number: needs calibration data
Segmentation (of chromosomes into constant copy number regions): an HMM-like algorithm
Analysing family CN data: a different HMMIncorporating non-polymorphic probes: independent HMM observations to be weighted and combined
Dealing with mixed normal-abnormal samplesUtilizing poor quality DNA samplesEstimating allele-specific copy number ……and more
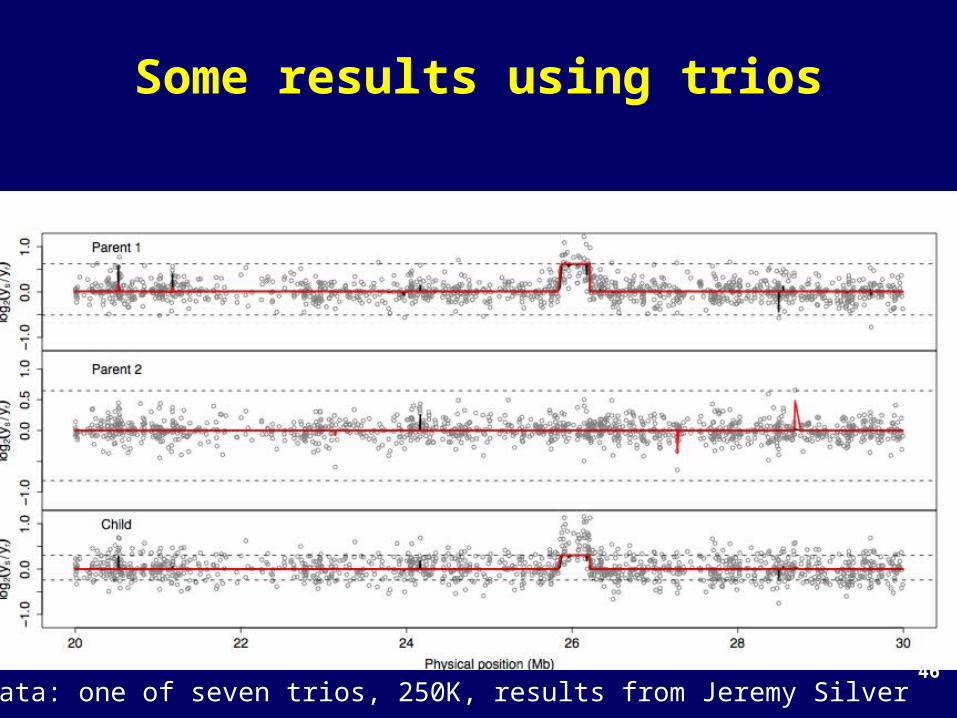
46
Some results using trios
Data: one of seven trios, 250K, results from Jeremy Silver

47
Conclusions/comments
• Using chromosome X permits us to:–test how well a method detects deletions–compare methods–get a sense of resolution
• We plan to do further tests with known CN changes to see how well this generalizes
• We are working on some of the issues other mentioned
There is room for contributions from you!

48
Available in aroma.affymetrix ("google it")
“Infinite” number of arrays: 1-1,000s
Requirements: 1-2GB RAMArrays: SNP, exon,
expression, (tiling).Dynamic HTML reportsImport/export to existing
methodsOpen source: RCross platform: Windows,
Linux, Mac

49
Acknowledgements
Henrik BengtssonHenrik Bengtsson, , UC BerkeleyUC Berkeley
Andrew Sinclair & Howard Slater,Andrew Sinclair & Howard Slater, MCRIMCRI
Nusrat RabbeeNusrat Rabbee, , GenentechGenentech
Simon Cawley, Francois Collin & Srinka Simon Cawley, Francois Collin & Srinka Ghosh,Ghosh, AffymetrixAffymetrix
Rafael Irizarry & Benilton CarvalhoRafael Irizarry & Benilton Carvalho, , Johns Johns HopkinsHopkins
Nancy ZhangNancy Zhang, , StanfordStanford
Jeremy SilverJeremy Silver, WEHI, WEHI

50
Thank you!


















