
02 farmacocinética
48
Dra. Floria Pancetti - UC N 2006 FARMACOCINÉTICA
-
Upload
daniel-alejandro -
Category
Travel
-
view
621 -
download
0
Transcript of 02 farmacocinética

Dra. Floria Pancetti - UCN 2006
FARMACOCINÉTICA

Dra. Floria Pancetti - UCN 2006
Farmacocinética:
Estudia el curso temporal de las concentraciones ycantidades de los fármacos, y de sus metabolitos, en loslíquidos biológicos, tejidos y excretas, así como su relación con la respuesta farmacológica, y construyemodelos adecuados para interpretar estos datos.

Dra. Floria Pancetti - UCN 2006
El fármaco interactúa con sus receptores en la biofase, para lo cual debe alcanzar un intervalo de concentraciones.
Si la concentración es muy baja: dosis sub-terapéutica
Si la concentración es muy alta: efectos tóxicos (indeseables)

Dra. Floria Pancetti - UCN 2006
La concentración de un fármaco en su lugar de accióndepende de la:
Absorción
Distribución
Eliminación

Dra. Floria Pancetti - UCN 2006
Fijación a tejidos inactivos
PlasmaTejidos activos
Metabolismo Excreción
D D x f F
FFP
FP
F M F M
M
F
M
M MP
PROCESOS FARMACOCINÉTICOS
Fex Mex

Dra. Floria Pancetti - UCN 2006
Curso temporalde la cantidad de fármaco en el lugar de absorción, del fármaco y su metabolito en elcuerpo, y delfármaco y sumetabolito excretadostras la administraciónde una dosis por víaextravascular.

Dra. Floria Pancetti - UCN 2006
Algunos conceptos:
Concentración mínima eficaz (CME): aquella porencima de la cual se observa el efecto terapéutico
Concentración mínima tóxica (CMT): aquella por encimade la cual suelen observarse efectos tóxicos.
Índice terapéutico: es el cociente entre CMT y CME
Periodo de latencia (PL): tiempo que transcurre desdela administración hasta el inicio del efecto (hasta que laconcentración plasmática alcanza la CME).

Dra. Floria Pancetti - UCN 2006
Intensidad del efecto: depende (para muchos fármacos)de la concentración máxima que se alcance.
Duración de la acción: también se conoce como tiempoeficaz (TE). Corresponde al tiempo transcurrido entreque se alcanza la CME y el momento en que desciendepor debajo de ésta.

Dra. Floria Pancetti - UCN 2006
Curso temporal de laconcentraciónplasmática de unfármaco y relación consus efectos.

Dra. Floria Pancetti - UCN 2006
Variabilidad individual
nº individuos
efec
to
Efecto deseado delfármaco
Ineficacia delfármaco
Efecto tóxico delfármaco

Dra. Floria Pancetti - UCN 2006
Factores que inciden en la variabilidad individual:
Factores fisiológicos: patrón genético, edad, hábitos alimenti- cios, ingesta de alcohol, hábito de fumar, embarazo.
Factores patológicos: función renal, hepática o cardiaca.
Interacciones con otros fármacos que alteren la respuesta.

Dra. Floria Pancetti - UCN 2006
Farmacocinética clínica:
Su objetivo es alcanzar y mantener la concentración plasmá-tica necesaria para conseguir el efecto terapéutico sin llegar a producir efectos tóxicos.

Dra. Floria Pancetti - UCN 2006
Mecanismos de transporte que inciden en los procesos farmacocinéticos
Moléculas pequeñas:
Difusión pasiva Difusión facilitada Transporte activo
Moléculas grandes:
Pinocitosis Exocitosis

Dra. Floria Pancetti - UCN 2006
Mecanismos de transporte que inciden en los procesos farmacocinéticos
Factores que inciden en la velocidad de difusión:
Tamaño Liposolubilidad Grado de ionización

Dra. Floria Pancetti - UCN 2006
Difusión pasiva:
La velocidad depende de la Ley de Fick:
A mayor gradiente de concentración, menor tamaño de la molécula y mayor liposolubilidad, mayor velocidad.
La mayoría de los fármacos son electrolitos débiles.Su nivel de ionización depende de su pKa y siguen lafórmula de Henderson-Hasselbach:
pH = pKa + log ([base]/[ácido])

Dra. Floria Pancetti - UCN 2006
Cuando la membrana separa los medios de diferente pH, elfármaco se acumulará en el lado donde se produzca sumayor nivel de ionización. Sin embargo nunca se produceun equilibrio debido a la continua absorción.

Dra. Floria Pancetti - UCN 2006

Dra. Floria Pancetti - UCN 2006
Transporte activo:
Funciona contra el gradiente electroquímico y requiere ATP.
Características:
Saturable
Susceptible de inhibición competitiva
Inhibido por mecanismos o substancias que interfieran con la producción de energía.
Ej.: túbulo proximal renal, tubo digestivo, tracto biliar, paso deLCR a la sangre, paso de sangre a la saliva.

Dra. Floria Pancetti - UCN 2006
Otros sistemas de transporte:
Filtración
Difusión facilitada
Exocitosis o endocitosis
Ionóforos
Liposomas

Dra. Floria Pancetti - UCN 2006
Filtración:Paso de sustancias a través de hendiduras intercelulares.
Difusión facilitada:Paso a sustancias a favor del gradiente de concentración através de proteínas transportadoras. Susceptible de saturacióne inhibición competitiva.
Exocitosis y endocitosis:Transporte de macromoléculas.
Ionóforos:Moléculas pequeñas sintetizadas por microorganismos que que aumentan la permeabilidad de la bicapa lipídica de la mem-brana.
Liposomas:Su usan para favorecer el acceso de fármaco a las células.Son estructuras sintéticas que forman bicapas.

Dra. Floria Pancetti - UCN 2006
ABSORCIÓN
Comprende las etapas de liberación del fármaco de suforma farmacéutica, su disolución, la entrada al organismodesde el lugar de administración, los mecanismos detransporte, la velocidad y la cantidad de fármaco queaccede a la circulación sistémica.
Depende de:
Las características fisicoquímicas del fármaco. Las características de la preparación farmacéutica Las características del lugar de absorción La eliminación presistémica y fenómeno “primer paso”

Dra. Floria Pancetti - UCN 2006
Primer paso hepático:
Se refiere a la metabolización hepática que se produce del fármaco absorbido en el tracto gastrointestinal y que llega al hígado por la vena porta antes de entrar en la circulación sistémica.

Dra. Floria Pancetti - UCN 2006

Dra. Floria Pancetti - UCN 2006
Vías de administraciónVías de administración
Vías enterales: oral, sublingual, rectal.
Vías parenterales: intravenosa, intraarterial, intramuscular,subcutánea.
Otras vías: dérmica, nasal, epidural, intratecal, intraventricular, inhalatoria, conjuntival, uretral, vesical, vaginal, intraperitoneal.

Dra. Floria Pancetti - UCN 2006
Cinética de absorciónCinética de absorción
Cuantifica la entrada del fármaco en la circulación sistémica.Contempla los estudios de la velocidad de absorción, lacantidad absorbida y los factores que la alteran.
Velocidad de absorción: número de moléculas de un fármacoque se absorbe por unidad de tiempo. Depende de laconstante de absorción (Ka) y del número de moléculas que seencuentren en solución en el lugar de absorción.
Semivida de absorción (t1/2a) : tiempo que tarda en reducirse a la mitad el número de moléculas que quedan por absorberse.

Dra. Floria Pancetti - UCN 2006
Absorción
Orden 1
Orden 0

Dra. Floria Pancetti - UCN 2006
Cantidad absorbida:
Es igual a la administrada cuando el fármaco se administra por vía intravascular y corresponde al área bajo la curva(AUC) de las concentraciones plasmáticas.
Otras vías: la cantidad absorbida es menor a la administradadebido a la preparación farmacéutica y a la eliminaciónpresistémica.

Dra. Floria Pancetti - UCN 2006
Fracción de absorción disponible (f):
Es la fracción de la dosis administrada que llega a la circulación sistémica en una forma inalterada. Se obtiene dividiendo el área bajo al curva obtenida tras la adminsitración extravascular (AUCev) por la obtenida por vía intravenosa (AUCiv), teniendo en cuenta la dosis administrada (D) por cada vía y el aclaramiento (Cl) del individuo.
f = AUCev
AUCiv
xDiv/(peso x Cliv)
Dev/(peso x Clev)

Dra. Floria Pancetti - UCN 2006
Cuando la comparación de las áreas se realiza en los mismospacientes y la dosis por vía extravascular es igual a laintravenosa la fórmula se simplifica a:
f = AUCev
AUCiv
La cantidad absorbida por vía extravascular será elproducto de la dosis administrada por la fracción de absorcióncorrespondiente a la forma farmacéutica y a la vía de administración utilizadas.
Cantidad absorbida = D x f

Dra. Floria Pancetti - UCN 2006
BiodisponibilidadBiodisponibilidad
La biodisponibilidad de un fármaco indica la velocidad y lacantidad de la forma inalterada de un fármaco que accede ala circulación sistémica y, por lo tanto, está disponible paraacceder a los tejidos y producir un efecto. Depende no sólo dela absorción, sino de la distribución y la eliminación.

Dra. Floria Pancetti - UCN 2006

Dra. Floria Pancetti - UCN 2006
Factores que alteran la absorción
Factores fisiológicos: En el reciénnacido, embarazo, ancianos, ingesta dealimentos.
Factores patológicos: La absorción oralpuede alterarse cuando hay vómitos, diarrea,enfermedades digestivas. Se ve alteradala absorción intramuscular por insuficienciacardiaca.
Factores yatrógenos: Interacciones que puedenafectar la absorción.

Dra. Floria Pancetti - UCN 2006
DISTRIBUCIÓN
La distribución de los fármacos permite su acceso a losórganos en los que debe actuar y a los órganos que lo van a eliminar y condiciona las concentraciones que alcanzan encada tejido.
Las moléculas de un fármaco son transportadas en la sangredisueltas en el plasma, fijadas a proteínas plasmáticas ounidas a células sanguíneas.
De todas estas posibilidades, la fijación a albúmina es la másfrecuente e importante. -glucoproteína es otra proteínaimportante para la unión, además de las lipoproteínas.

Dra. Floria Pancetti - UCN 2006
Distribución en los tejidos
Distribución regional:
Fármacos liposolubles: acceso fácil a órganos irrigados(cerebro, corazón, hígado, riñones).
Fármacos menos liposolubles: acceso fácil a tejidos cuyoscapilares son ricos en hendiduras intercelulares (sinusoideshepáticos).
La mayoría de los fármacos tienen la capacidad de fijarse adeterminados tejidos en los que alcanzan concentracionesmás altas que en el resto del organismo. Ej.: acumulación defármacos liposolubles en la grasa.

Dra. Floria Pancetti - UCN 2006
Distribución a áreas especiales:
SNC Ojo Circulación fetal Acceso a secreciones exocrinas (lágrimas, saliva, leche o líquido prostático)
Difusión pasiva o transporte activo

Dra. Floria Pancetti - UCN 2006
Barrera hematoencefálica (BHE):
Formada por un conjunto de estructuras que limitanel paso de sustancias hidrófilas desde los capilares hasta elSNC. las células endoteliales de los capilares están íntima-mente adosadas sin dejar espacios intercelulares debido ala presencia de zona ocludens. Además hay una membranabasal que forma un revestimiento continuo alrededor delendotelio. Las prolongaciones de los astrocitos tambiénrevisten la superficie capilar.
Los fármacos sólo atraviesan la BHE por difusiónpasiva. En algunos casos hay transporte activo (hexosas,aminoácidos, ácidos monocarboxílicos de cadena corta).
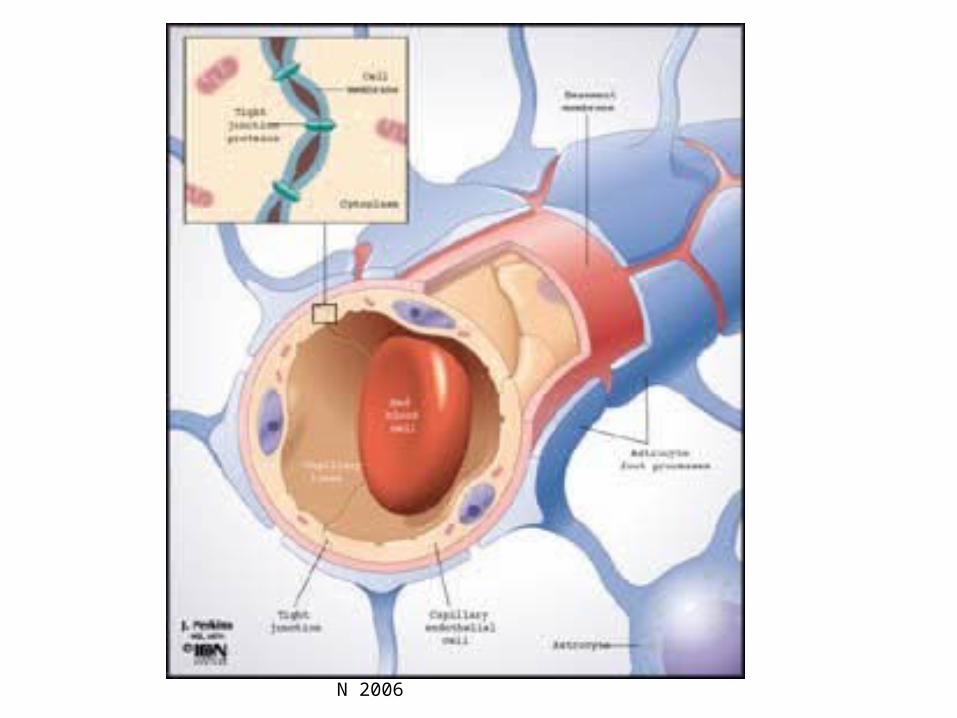
Dra. Floria Pancetti - UCN 2006

Dra. Floria Pancetti - UCN 2006
Existe metabolización en células endoteliales de fármacos:L-dopa dopamina.
Algunos núcleos cerebrales carecen de BHE, por lo tanto sefavorece el ingreso de fármacos en esos puntos.Ej: eminencia media, área postrema, órgano subfornical, glándula pineal, órgano subcomisural.
Condiciones patológicas que alteran la permeabilidad de laBHE: isquemia, anoxia, traumatismos, neoplasias, infecciones,enfermedades autoinmunes, hipertensión intracraneal.

Dra. Floria Pancetti - UCN 2006
Barrera placentaria:
Separa a la madre y el feto. Para atravesarla, los fármacos y sus metabolitos tiene que salir de los capilares maternos, atravesar una capa de células trofoblásticas y mesenquimáticasy entrar en los capilares fetales.
Los fármacos traspasan por difusión pasiva.
La placenta tiene enzimas que pueden metabolizar los fármacos.

Dra. Floria Pancetti - UCN 2006
Compartimentos farmacocinéticos
Compartimento central: incluye agua plasmática, intersticial e intracelular de los órganos bien irrigados y SNC si el fármacoatraviesa la BHE.
Compartimento periférico superficial: agua intracelular pocoaccesible de los órganos poco irrigados (piel, grasa, músculo omédula ósea.
Compartimento periférico profundo: depósitos tisulares a los queel fármaco se une fuertemente y de los cuales se libera conlentitud.

Dra. Floria Pancetti - UCN 2006

Dra. Floria Pancetti - UCN 2006

Dra. Floria Pancetti - UCN 2006
ELIMINACIÓN
La concentración activa del fármaco disminuye por lametabolización y la excreción.
Sistema de excreción según su importancia:
Vía urinaria > vía biliar-entérica > sudor > saliva > leche > epitelios descamados
Vía urinaria: la cantidad final de un fármaco que se excreta porla orina es la resultante de la filtración glomerular y de la secreción tubular, menos la reabsorción tubular.

Dra. Floria Pancetti - UCN 2006
Excreción biliar e intestinal: circulación enterohepática:
La excreción biliar está muy relacionada con los procesos debiotransformación. Se produce principalmente por secreciónactiva con diferentes sistemas de transporte para sustanciasácidas, básicas y neutras. Se eliminan principalmente porla bilis:
Sustancias con elevado peso molecularSustancias con grupos polaresCompuestos no ionizablesAlgunos compuestos organometálicos

Dra. Floria Pancetti - UCN 2006
Excreción intestinal:
Los fármacos pueden pasar directamente de la sangre a la luz intestinal, por difusión pasiva, en partes distales en que elgradiente de concentración y la diferencia de pH lo favorezcan.
Circulación enterohepática:
Los fármacos eliminados a la luz intestinal en forma activa através de la bilis o del epitelio intestinal pueden reabsorberse pasivamente en el intestino a favor de un gradiente deconcentración. Este fenómeno prolonga la duración del efecto.

Dra. Floria Pancetti - UCN 2006
Excreción a la leche:
Los fármacos pasan a la leche por difusión pasiva en formaproporcional a su liposolubilidad e indirectamente proporcionalsegún su grado de ionización y unión a proteínas plasmáticas.
Excreción a la saliva:
Poco importante desde el punto de vista cuantitativo. Ademásla mayor parte del fármaco excretado por la saliva pasa al tubodigestivo donde es reabsorbido.

Dra. Floria Pancetti - UCN 2006
Aclaramiento (clearence):
El aclaramiento (Cl) de un fármaco por un órgano indica lacapacidad de ese órgano para eliminarlo. Se expresamediante el número de mililitros de plasma que el órgano aclara (es decir, de los que elimina totalmente el fármaco) porunidad de tiempo.

Dra. Floria Pancetti - UCN 2006


















